Tryptophan and Cognitive Function
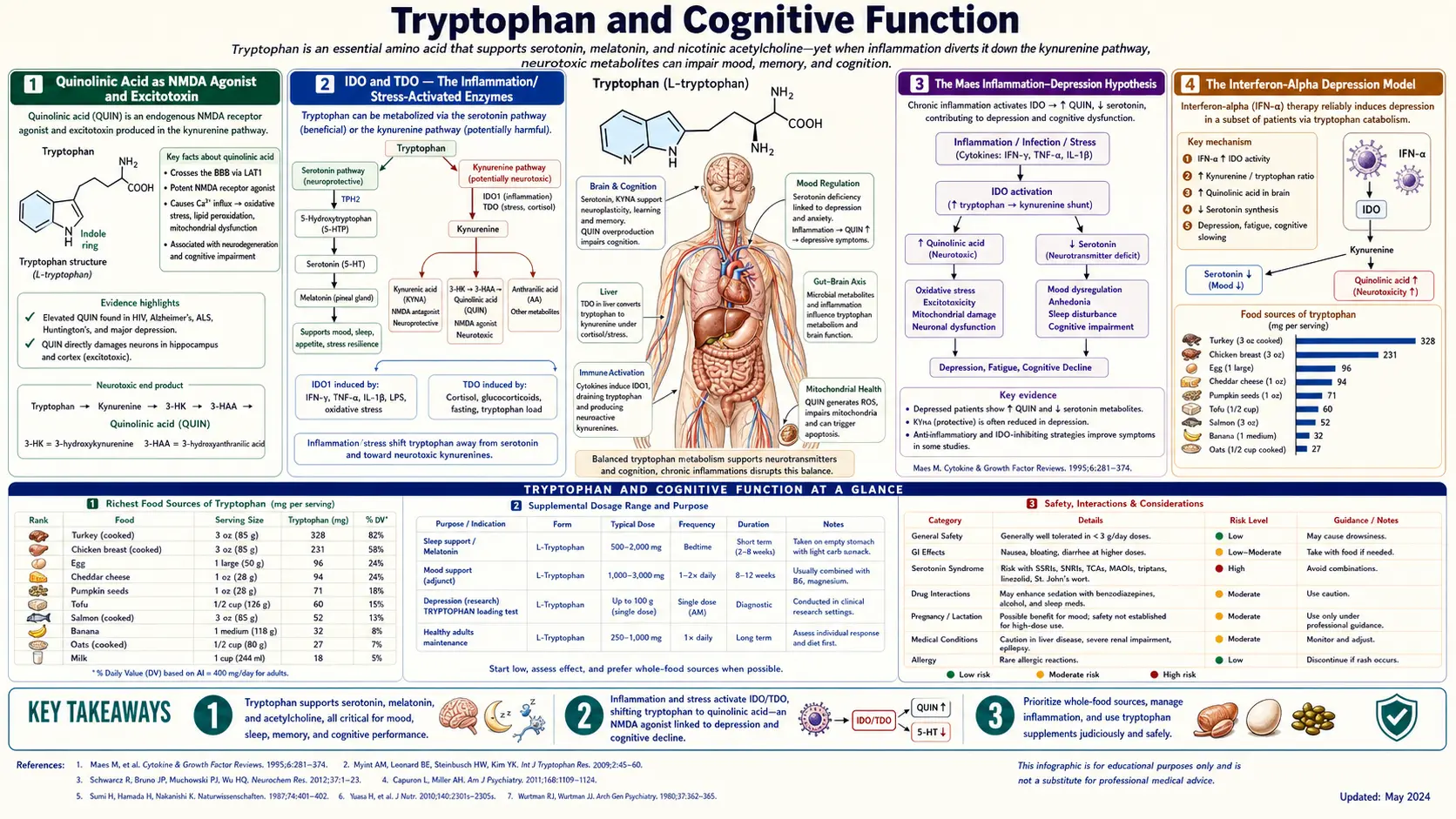
Tryptophan's most consequential effect on cognitive function does not come from serotonin biology, but from the kynurenine pathway and its excitotoxic metabolite quinolinic acid. Quinolinic acid is an NMDA glutamate receptor agonist and a generator of reactive nitrogen species; in the inflamed brain it accumulates to neurotoxic concentrations and contributes to neuronal injury in major depression, suicidal ideation, schizophrenia, Alzheimer's disease, Huntington's disease, HIV-associated neurocognitive disorder, and chronic interferon-alpha therapy. The rate-limiting enzymes IDO (indoleamine 2,3-dioxygenase) and TDO (tryptophan 2,3-dioxygenase) are induced by inflammation (interferon-gamma) and stress (cortisol), respectively — making the kynurenine pathway one of the most important molecular bridges between systemic inflammation, chronic stress, depression, and neurodegeneration. Michael Maes formalized this in the inflammation-depression hypothesis in the 1990s, and the field has since produced an enormous body of clinical evidence linking interferon-induced depression, autoimmune-associated depression, and CSF quinolinic-to-kynurenic ratios as candidate biomarkers. The kynurenic acid branch acts as the neuroprotective counterbalance: kynurenic acid is an NMDA antagonist and is broadly neuroprotective. This page walks through quinolinic acid as an NMDA agonist and excitotoxin, the IDO/TDO regulatory biology, the Maes inflammation-depression hypothesis, the kynurenic-acid neuroprotection counterweight, and the implications for cognition, mood, and neurodegenerative disease.
Table of Contents
- Quinolinic Acid as NMDA Agonist and Excitotoxin
- IDO and TDO — The Inflammation/Stress-Activated Enzymes
- The Maes Inflammation-Depression Hypothesis
- The Interferon-Alpha Depression Model
- Quinolinic Acid and Suicidal Ideation
- Quinolinic Acid in Alzheimer's Disease
- Huntington's Disease and the Striatum
- The Schizophrenia Paradox — Elevated Kynurenic Acid
- Kynurenic Acid as Neuroprotective Counterbalance
- KMO Inhibitors as Drug Targets
- Practical Cognitive Implications
- Key Research Papers
- Connections
- Featured Videos
Quinolinic Acid as NMDA Agonist and Excitotoxin
Quinolinic acid (2,3-pyridinedicarboxylic acid, often abbreviated QUIN) is the penultimate metabolite of the kynurenine pathway before the committing step to NAD+ biosynthesis. Under normal physiologic conditions, brain quinolinic acid concentrations are low (nanomolar to low-micromolar range). Under chronic inflammation, microglial activation, or persistent interferon signaling, quinolinic acid concentrations can rise to micromolar concentrations that produce two simultaneous neurotoxic effects:
- NMDA receptor agonism — quinolinic acid is a selective agonist at the NMDA subtype of glutamate ionotropic receptors. NMDA receptors are calcium-permeable channels that, when activated chronically or excessively, produce intracellular calcium overload, mitochondrial dysfunction, generation of reactive oxygen and nitrogen species, and ultimately neuronal death via excitotoxicity. The neurons most vulnerable to NMDA-mediated excitotoxicity are the medium spiny neurons of the striatum, hippocampal CA1 pyramidal neurons, and pyramidal neurons of the cortex — precisely the regions affected in Huntington's disease and in late Alzheimer's disease.
- Free radical generation — quinolinic acid forms a complex with iron and catalyzes the generation of hydroxyl radicals via Fenton-like chemistry. It also stimulates nitric oxide synthase induction in adjacent astrocytes, generating peroxynitrite (the highly reactive product of nitric oxide combining with superoxide). The combination produces oxidative and nitrosative stress that extends the neurotoxic effect beyond direct NMDA-receptor channels.
The clinical-pharmacology evidence for quinolinic acid as a true neurotoxin (rather than a passive biomarker) comes from animal models: direct intrastriatal injection of quinolinic acid in rats produces a precise excitotoxic lesion that is the standard rodent model of Huntington's disease. The lesion preferentially destroys striatal medium spiny neurons while sparing cholinergic interneurons — exactly the pattern seen in Huntington's pathology.
For practical context: typical brain quinolinic acid concentrations in healthy adults are 30–100 nM. In severe inflammation, sepsis, advanced HIV-associated neurocognitive disorder, or active interferon-alpha therapy, concentrations can rise 10–100 fold to the low-micromolar range. These elevations correlate with clinical neurocognitive impairment and depressive symptoms.
IDO and TDO — The Inflammation/Stress-Activated Enzymes
The kynurenine pathway has two rate-limiting enzymes that catalyze the same chemical reaction (oxidation of tryptophan's indole ring at the C2-C3 bond) but are regulated by completely different signals:
- Indoleamine 2,3-dioxygenase 1 (IDO1) — expressed in most peripheral tissues including the lung, gut, placenta, dendritic cells, macrophages, and microglia. The principal inducer is interferon-gamma, the master cytokine of Th1 cellular immunity. IFN-gamma can raise IDO1 expression 100-fold within hours. Other inflammatory cytokines (IFN-alpha, TNF-alpha, IL-1, IL-6) also contribute. IDO1 is also induced in tumor cells as part of immune escape biology — tumors that upregulate IDO1 deplete local tryptophan and starve attacking T cells.
- Indoleamine 2,3-dioxygenase 2 (IDO2) — a related enzyme with more restricted expression. Less well-characterized than IDO1.
- Tryptophan 2,3-dioxygenase (TDO) — expressed predominantly in the liver. Induced by cortisol and by high substrate (tryptophan) availability. TDO is the dominant enzyme handling postprandial tryptophan load and is the principal route for tryptophan disposition in unstressed metabolism.
The regulatory architecture means that any state of systemic inflammation or chronic psychological stress can shunt dietary tryptophan into the kynurenine pathway, with two downstream consequences:
- Less tryptophan available for serotonin/melatonin synthesis — predisposing to depression and insomnia.
- More tryptophan flowing into the kynurenine pathway, with the inflammation-specific bias toward the quinolinic-acid (neurotoxic) rather than the kynurenic-acid (neuroprotective) branch — predisposing to cognitive impairment and neurodegeneration.
This is the molecular pivot that explains why inflammation, chronic stress, depression, and cognitive decline are so consistently clustered in clinical observation.
The Maes Inflammation-Depression Hypothesis
Michael Maes, a Belgian psychiatrist, was one of the earliest and most persistent proponents of the now-mainstream view that major depression is, in many cases, fundamentally an inflammatory disorder. Beginning in the early 1990s, Maes and colleagues published a series of papers documenting that depressed patients had:
- Elevated serum inflammatory cytokines (IL-1, IL-6, TNF-alpha, sIL-2R, acute-phase proteins).
- Elevated indices of oxidative and nitrosative stress (lipid peroxides, oxidized LDL, reduced glutathione, decreased antioxidant capacity).
- Activated indoleamine 2,3-dioxygenase with reduced plasma tryptophan and shifted tryptophan/competing-amino-acid ratios.
- Elevated kynurenine and quinolinic-acid metabolites in plasma and CSF.
- Increased intestinal permeability ("leaky gut") with elevated antibodies to bacterial LPS components, contributing to chronic low-grade systemic inflammation.
Maes integrated these findings into what he called the IO&NS hypothesis (Inflammation and Oxidative & Nitrosative Stress). In this framework, depression is not simply a serotonin-deficiency disorder but is the brain's response to chronic peripheral inflammation, which acts through kynurenine pathway activation, microglial priming, oxidative stress, and reduced serotonin synthesis to produce the clinical syndrome of major depression.
The implications include:
- Anti-inflammatory interventions should have antidepressant effects (and several do — omega-3 fatty acids, curcumin, and aspirin have all shown modest antidepressant signals in trials).
- Medical conditions associated with chronic inflammation (rheumatoid arthritis, inflammatory bowel disease, chronic hepatitis, cardiovascular disease, type 2 diabetes) should have elevated rates of comorbid depression — and they do.
- Inflammatory biomarkers should predict treatment response — emerging evidence suggests that elevated CRP and IL-6 predict poorer response to SSRI monotherapy and better response to certain anti-inflammatory adjuncts.
- Treatments targeting the kynurenine pathway (KMO inhibitors, IDO inhibitors) should have antidepressant potential — this is an active drug-development area.
The Maes IO&NS framework has become increasingly mainstream in academic psychiatry over the past fifteen years, particularly with the rise of the broader "inflammation in psychiatry" movement led by figures like Andrew Miller at Emory and Charles Raison.
The Interferon-Alpha Depression Model
The cleanest clinical proof-of-concept for the inflammation-kynurenine-depression hypothesis comes from the iatrogenic depression produced by therapeutic interferon-alpha. From the 1990s through the 2010s, interferon-alpha was widely used to treat chronic hepatitis C, certain hematologic malignancies (CML, hairy cell leukemia), and malignant melanoma. The treatment was famously effective at suppressing viral replication but carried a striking neuropsychiatric side-effect profile: up to 30% of patients receiving sustained IFN-alpha therapy develop a depressive syndrome that meets DSM criteria for major depressive disorder, often emerging within weeks to months of treatment initiation.
This iatrogenic depression has been one of the most studied human models of cytokine-induced depression. Key findings:
- IFN-alpha rapidly activates IDO and drives plasma tryptophan down by 30–50% within weeks.
- CSF quinolinic acid rises measurably and correlates with depression-rating-scale scores.
- The kynurenine/tryptophan ratio in plasma rises substantially and correlates with severity of depressive symptoms.
- SSRI pretreatment with paroxetine (Capuron, Miller, et al.) reduces the incidence of IFN-alpha-induced major depression from ~30% to under 10%, supporting a causal role for serotonin depletion downstream of kynurenine pathway activation.
- Symptoms resolve over weeks to months after IFN-alpha discontinuation, paralleling the recovery of plasma tryptophan and the resolution of the kynurenine pathway activation.
With the rise of direct-acting antiviral agents for hepatitis C, IFN-alpha is no longer the standard of care, but the model remains scientifically important. It is the strongest available evidence that a controllable, time-limited cytokine stimulus can cause major depression in previously well humans, and that the kynurenine pathway is mechanistically central to the connection.
Quinolinic Acid and Suicidal Ideation
One of the most striking lines of contemporary kynurenine pathway research is the work of Lena Brundin, Sophie Erhardt, and colleagues at the Karolinska Institute on quinolinic acid and suicidality. In a series of papers beginning around 2009 and culminating in major publications in 2013 and after, the group demonstrated:
- Patients hospitalized after recent suicide attempts had two- to three-fold elevations in CSF quinolinic acid compared to non-attempting depressed controls and to healthy controls.
- The elevation was largely independent of depression severity scores and appeared to be a more specific biomarker of suicidality than of depression itself.
- Quinolinic acid concentrations normalized over weeks to months of clinical recovery, paralleling reduced suicidal ideation.
- The elevation was accompanied by reduced CSF kynurenic acid in some studies, magnifying the kynurenic / quinolinic ratio shift toward the neurotoxic side.
- Plasma kynurenine metabolite ratios correlated with the CSF findings, suggesting peripheral biomarkers could be developed.
The hypothesis emerging from this work is that quinolinic acid-driven NMDA receptor overactivation in cortical and limbic regions may contribute to the neurobiology of suicidal ideation specifically — over and above the more general inflammation-depression link. This is biologically interesting because it dovetails with the discovery that NMDA receptor antagonists like ketamine produce rapid antidepressant and anti-suicidal effects. If suicidality is driven in part by NMDA receptor hyperactivity from elevated quinolinic acid, then antagonizing NMDA receptors should produce rapid relief — and ketamine does. The kynurenine biology and the ketamine biology may be two sides of the same molecular coin.
Quinolinic Acid in Alzheimer's Disease
The kynurenine pathway is now broadly recognized as one of the inflammatory contributors to Alzheimer's disease pathology. Multiple lines of evidence have accumulated:
- Quinolinic acid concentrations are elevated in the cortex and hippocampus of postmortem Alzheimer's brain tissue, particularly in regions adjacent to amyloid plaques where microglial activation is most intense.
- The kynurenine pathway intermediates are detectable in cerebrospinal fluid and the kynurenine/tryptophan ratio is elevated in Alzheimer's patients compared to age-matched controls.
- Inflammatory cytokines that activate IDO (IFN-gamma, IL-1-beta, TNF-alpha) are reliably elevated in Alzheimer's brain and CSF.
- Microglia surrounding amyloid plaques express high levels of IDO and produce quinolinic acid locally.
The integrative picture is that chronic neuroinflammation — whether triggered by amyloid-beta accumulation, tau pathology, or peripheral inflammatory contributors — activates the microglial kynurenine pathway, generates local quinolinic acid, and contributes to excitotoxic neuronal injury that compounds the primary amyloid/tau pathology. This is not the cause of Alzheimer's disease in the strict sense; it is a contributing mechanism of the neurodegenerative cascade. It is also one of the rationales for ongoing interest in anti-inflammatory and microglial-targeted therapies for Alzheimer's disease.
Huntington's Disease and the Striatum
Huntington's disease is the textbook example of selective excitotoxicity. The disease is caused by an expanded CAG trinucleotide repeat in the huntingtin gene, producing mutant huntingtin protein that accumulates in neurons. The cellular pathology preferentially affects the medium spiny neurons of the striatum — the projection neurons of the caudate and putamen. The progressive striatal atrophy is the anatomic basis of the motor (chorea) and cognitive (executive dysfunction, dementia) manifestations of the disease.
The striking observation in the kynurenine literature is that direct intrastriatal injection of quinolinic acid in rats produces a precise lesion that recapitulates the cellular pathology of Huntington's disease. Medium spiny neurons die selectively while cholinergic interneurons are spared. This quinolinic-acid lesion model has been the standard rodent model of Huntington's disease for decades.
In human Huntington's disease tissue, kynurenine pathway enzymes are upregulated in microglia, kynurenine pathway metabolites are altered (with kynurenic acid initially elevated and then depleted as the disease progresses), and the kynurenic / quinolinic ratio shifts toward neurotoxicity over time. The relevance of the kynurenine pathway to Huntington's pathogenesis is now well established, and KMO inhibitors that shunt flux toward the kynurenic (protective) branch have shown promise in animal models of the disease.
The Schizophrenia Paradox — Elevated Kynurenic Acid
Schizophrenia presents an interesting paradox in kynurenine biology. While most psychiatric and neurodegenerative conditions show increased quinolinic acid relative to kynurenic acid, several studies have found that schizophrenia patients have elevated kynurenic acid concentrations in CSF and postmortem brain tissue. This is biologically meaningful because kynurenic acid is a NMDA receptor antagonist — and the leading neurobiological hypothesis of schizophrenia is that NMDA receptor hypofunction contributes to the disorder's positive, negative, and cognitive symptoms.
The schizophrenia evidence base includes:
- Increased CSF kynurenic acid in male schizophrenia patients compared to controls (Erhardt et al. 2001).
- Increased kynurenic acid in postmortem prefrontal cortex of schizophrenia patients.
- Genetic variants in kynurenine pathway enzymes associated with schizophrenia risk.
- Animal models in which elevated brain kynurenic acid produces deficits in prepulse inhibition, working memory, and social interaction — behaviors that translate to schizophrenia-relevant phenotypes.
This is one of the threads connecting the dissociative anesthetic NMDA antagonists (ketamine, PCP) to schizophrenia-like states — both pharmacologic NMDA antagonism and endogenous elevation of the natural NMDA antagonist kynurenic acid can produce overlapping symptomatology. It also raises the interesting possibility that different psychiatric disorders may correspond to different points on the kynurenic-quinolinic continuum: depression and suicidality at the quinolinic (excitotoxic) end, schizophrenia at the kynurenic (NMDA-hypofunction) end.
Kynurenic Acid as Neuroprotective Counterbalance
Kynurenic acid is the kynurenine pathway's good news. Produced by the kynurenine aminotransferases (KAT I, KAT II, KAT III, KAT IV), all of which require pyridoxal-5-phosphate as cofactor, kynurenic acid acts at multiple receptors with broadly protective effects:
- NMDA receptor antagonism — binds the glycine modulatory site of the NMDA receptor and reduces glutamatergic neurotransmission. This is the principal neuroprotective mechanism.
- Alpha-7 nicotinic acetylcholine receptor antagonism — reduces presynaptic glutamate release and modulates cognitive function.
- GPR35 agonism — an orphan G-protein-coupled receptor with anti-inflammatory effects, particularly in the gut and immune cells.
- Free radical scavenging — kynurenic acid is a modest direct antioxidant and reduces oxidative stress in inflamed tissue.
Astrocytes are the principal producers of kynurenic acid in the brain. Astrocytes express KAT II at high levels and continuously produce kynurenic acid as part of their tonic neuromodulation of synaptic glutamate. In acute injury (stroke, traumatic brain injury), astrocyte kynurenic acid production rises as part of the endogenous neuroprotective response.
The therapeutic implication is that boosting kynurenic acid (or its synthetic precursors that cross the BBB more readily than kynurenic acid itself) might be neuroprotective in stroke, traumatic brain injury, multiple sclerosis, and excitotoxic neurodegenerative disease. Several research programs are pursuing this strategy.
KMO Inhibitors as Drug Targets
Kynurenine 3-monooxygenase (KMO) is the enzyme at the kynurenic-quinolinic branchpoint. It directs kynurenine flux toward the quinolinic-acid (neurotoxic) branch. Inhibiting KMO has the dual effect of:
- Reducing production of quinolinic acid downstream — reducing excitotoxic burden.
- Shunting more kynurenine into the kynurenic-acid (neuroprotective) branch — raising endogenous neuroprotection.
KMO inhibitors are an active drug-discovery area in pharmaceutical research. Several have entered preclinical and early clinical development for Huntington's disease, Alzheimer's disease, major depression, and inflammatory disorders. The class's pharmacological appeal is that it intervenes at a regulatable branchpoint rather than at any single downstream target, potentially producing broad effects with a single pharmacologic action.
Brain-penetrant KMO inhibitors have been the major design challenge — many candidate molecules have good potency in vitro but poor blood-brain barrier penetration, limiting their effect to peripheral kynurenine pathway modulation. The peripheral effect alone produces measurable changes in brain kynurenine pathway metabolites (because circulating kynurenine itself crosses the BBB and shifts brain pathway dynamics), but the peripheral-only constraint has slowed development.
Adjacent drug classes that target the same axis include IDO1 inhibitors (epacadostat and others, mainly in oncology trials), TDO inhibitors (less clinically advanced), and AhR modulators (the aryl hydrocarbon receptor is a kynurenine-activated transcription factor with broad immune and metabolic effects).
Practical Cognitive Implications
For everyday clinical practice, the kynurenine pathway biology translates into several practical implications for patients concerned with cognitive function, mood, and long-term brain health:
- Chronic inflammation is bad for brain function in part through the kynurenine pathway. The general advice to reduce chronic inflammatory burden — weight management, regular physical activity, anti-inflammatory dietary patterns (Mediterranean diet, omega-3 fatty acids, polyphenol-rich foods, fiber), adequate sleep, stress management — has a specific molecular justification in kynurenine biology and is not just generic wellness advice.
- Supplemental tryptophan in active inflammation may not produce the expected serotonin boost because the substrate is being shunted into kynurenine. Resolving the underlying inflammation should come first.
- Cofactor adequacy is essential. Vitamin B6 (active form pyridoxal-5-phosphate) is required for both kynureninase (kynurenine pathway) and AADC (serotonin pathway) and for the kynurenine aminotransferases that produce protective kynurenic acid. Vitamin B2 (riboflavin) is required for KMO. Iron is required for IDO/TDO. Suboptimal status of any of these can distort the pathway in unhelpful directions.
- Major depression with prominent cognitive features and/or inflammation should prompt consideration of the inflammatory-depression subtype. Elevated CRP, IL-6, or other inflammatory markers may suggest a poorer response to SSRI monotherapy and a better response to anti-inflammatory adjuncts (omega-3 fatty acids, curcumin, NSAIDs in some studies, or in research settings IDO/KMO inhibitors as they become available).
- Patients with chronic inflammatory disease (rheumatoid arthritis, IBD, hepatitis C, type 2 diabetes, cardiovascular disease) should be actively screened for depression and cognitive symptoms, because the molecular biology predicts elevated risk and routine screening catches early cases that benefit from intervention.
- Brain fog in chronic illness has a kynurenine-pathway interpretation. The combination of low serotonin tone (substrate diversion) and elevated quinolinic acid (microglial activation) produces the typical clinical picture of dampened mood, reduced cognitive flexibility, and difficulty concentrating that patients describe. See Brain Fog.
Key Research Papers
- Maes M, Berk M, Goehler L, et al. (2012). Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Medicine 10:66. — PubMed
- Capuron L, Miller AH (2011). Immune system to brain signaling: neuropsychopharmacological implications. Pharmacology & Therapeutics 130:226–238. — PubMed
- Erhardt S, Lim CK, Linderholm KR, et al. (2013). Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology 38:743–752. — PubMed
- Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ (2012). Kynurenines in the mammalian brain: when physiology meets pathology. Nature Reviews Neuroscience 13:465–477. — PubMed
- Capuron L et al. (2002). Treatment of cytokine-induced depression. Brain Behavior and Immunity. — PubMed
- Wonodi I et al. (2011). Downregulated kynurenine 3-monooxygenase gene expression and enzyme activity in schizophrenia and bipolar disorder. Archives of General Psychiatry 68:665–674. — PubMed
- Beal MF et al. (1986). Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature 321:168–171. — PubMed
- Guillemin GJ, Brew BJ (2002). Implications of the kynurenine pathway and quinolinic acid in Alzheimer's disease. Redox Report 7:199–206. — PubMed
- Heyes MP et al. (1992). Quinolinic acid in cerebrospinal fluid and serum in HIV-1 infection: relationship to clinical and neurological status. Annals of Neurology. — PubMed
- Bonaccorso S et al. (2002). Increased depressive ratings in patients with hepatitis C receiving interferon-alpha-based immunotherapy are related to interferon-alpha-induced changes in the serotonergic system. Journal of Clinical Psychopharmacology. — PubMed
- Stone TW, Darlington LG (2002). Endogenous kynurenines as targets for drug discovery and development. Nature Reviews Drug Discovery 1:609–620. — PubMed
- Sundaram G, Brew BJ et al. (2018). The kynurenine pathway: a new metabolic axis in chronic depression. Neurochemistry International. — PubMed
PubMed Topic Searches
- PubMed: Kynurenine pathway and depression
- PubMed: Quinolinic acid neurotoxicity
- PubMed: Kynurenic acid neuroprotection
- PubMed: IDO induction by inflammation
- PubMed: KMO inhibitors
Connections
- Tryptophan Overview
- Tryptophan Benefits Hub
- Tryptophan for Sleep
- Tryptophan for Mood
- Tryptophan and Niacin
- Depression
- Anxiety
- Brain Fog
- Vitamin B6 (Multiple Pathway Enzymes)
- Vitamin B3 (Niacin / NAD+)
- Omega-3 Fatty Acids (Anti-Inflammatory)
- Gut-Brain Axis
- Inflammatory Bowel Disease
- Iron (IDO/TDO Cofactor)
- All Amino Acids
- Schizophrenia — the kynurenine paradox: elevated kynurenic acid and NMDA receptor hypofunction.