Asparagine for Protein Synthesis and N-Glycosylation
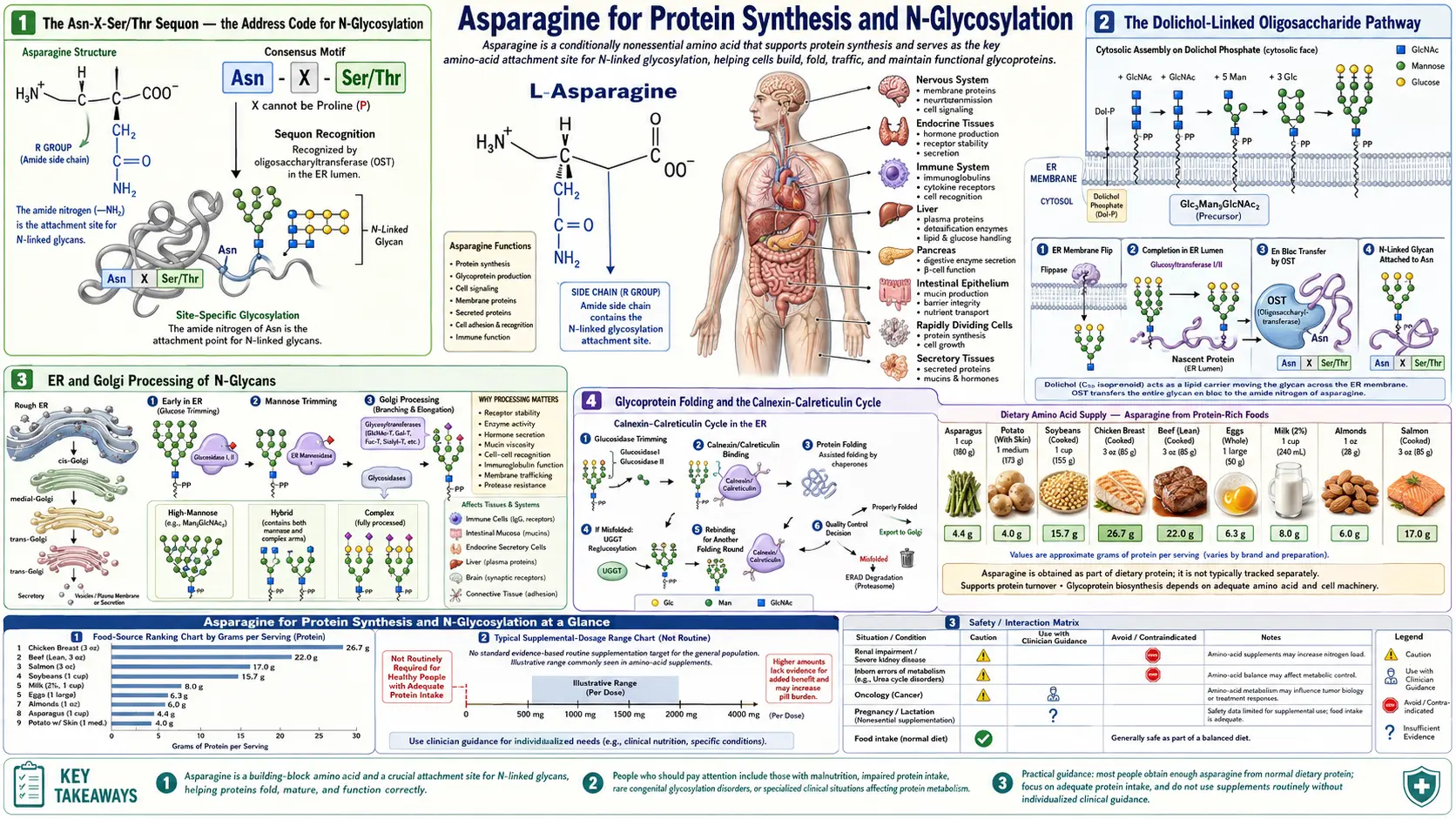
Virtually every secreted protein and every membrane-bound protein in the human body carries sugar chains attached to specific asparagine residues. This decoration — N-linked glycosylation — is not optional cellular ornamentation. It is the post-translational modification that allows the cell to fold its proteins correctly, target them to the right destinations, and protect them from premature proteolytic degradation. The reaction is catalyzed by the oligosaccharyltransferase (OST) complex in the rough endoplasmic reticulum, which transfers a pre-built 14-sugar glycan en bloc from a dolichol lipid carrier to the side-chain amide nitrogen of an asparagine residue, but only when that asparagine sits inside the consensus sequon Asn-X-Ser/Thr (where X is any amino acid except proline). The Asn-X-Ser/Thr sequon appears, on average, every 67 residues in the human proteome — meaning a typical secretory protein carries between 2 and 20 N-glycosylation sites. When the machinery breaks, the consequences are catastrophic: the family of congenital disorders of glycosylation (CDG) now numbers more than 150 distinct genetic conditions, most presenting in infancy with multisystem disease that maps directly onto the function of the glycoproteins that failed to assemble correctly.
Table of Contents
- The Asn-X-Ser/Thr Sequon — the Address Code for N-Glycosylation
- The Dolichol-Linked Oligosaccharide Pathway
- ER and Golgi Processing of N-Glycans
- Glycoprotein Folding and the Calnexin-Calreticulin Cycle
- Immunoglobulin Folding and the IgG-Fc Glycan
- Congenital Disorders of Glycosylation (CDG)
- Secreted Proteins, Membrane Proteins, and Cell-Surface Receptors
- Other Post-Translational Roles of Asparagine
- Asparagine Deamidation and the Molecular Clock of Aging
- Cautions and Clinical Considerations
- Key Research Papers
- Connections
- Featured Videos
The Asn-X-Ser/Thr Sequon — the Address Code for N-Glycosylation
N-linked glycosylation is the most common post-translational modification of eukaryotic secreted and membrane proteins. The site of attachment is the side-chain amide nitrogen of an asparagine residue, and the glycan is a branched chain of 14 sugars (Glc3-Man9-GlcNAc2) preassembled on a dolichol-phosphate lipid carrier in the endoplasmic reticulum membrane.
Crucially, not every asparagine is glycosylated. The oligosaccharyltransferase (OST) complex recognizes a specific tripeptide motif — the sequon — consisting of asparagine followed by any amino acid except proline, followed by either serine or threonine. The recognition rules:
- Position +1: any of the 20 amino acids except proline (proline at +1 sterically distorts the geometry required for OST recognition)
- Position +2: must be serine or threonine; the hydroxyl group of these residues participates in a hydrogen-bonding network that activates the asparagine amide nitrogen as a nucleophile
- Accessibility: the sequon must be exposed on the surface of the nascent polypeptide as it threads through the OST active site — sequons buried in already-folded domains are skipped
The Asn-X-Ser/Thr sequon appears, by chance, every 67 residues on average in the human proteome. A typical secretory protein of 400 residues thus contains 3 to 8 sequons, and depending on accessibility, 1 to 6 of them are actually glycosylated in the mature protein. Some proteins are unusually glycan-dense: immunoglobulin G has 2 N-glycans on the Fc fragment plus variable numbers on the Fab; erythropoietin has 3 N-glycans on a 165-residue mature protein; the human chorionic gonadotropin beta subunit has 4 N-glycans.
The biological consequences of N-glycosylation are profound:
- Folding — the glycan acts as a recognition tag for the calnexin/calreticulin chaperone system, ensuring that only properly folded proteins exit the ER
- Targeting — mannose-6-phosphate tags on certain glycoproteins direct them to lysosomes via the M6P receptor
- Solubility — glycans dramatically increase the solubility and reduce the aggregation propensity of secreted proteins
- Half-life — terminally sialylated glycans protect circulating proteins from clearance by the hepatic asialoglycoprotein receptor; loss of sialic acid (the "Ashwell-Morell receptor" recognition signal) leads to rapid hepatic clearance
- Function — glycans on receptors modulate ligand binding, on enzymes modulate activity, on adhesion molecules modulate cell-cell interactions
- Immune evasion — HIV envelope glycoproteins, SARS-CoV-2 spike, and other viral glycoproteins use heavy N-glycan shielding to hide protein epitopes from antibody recognition
The cell's reliance on asparagine for this central modification is part of why ASNS expression rises so steeply in any cell that is upregulating protein synthesis — whether that is a B cell secreting antibody, a hepatocyte producing albumin, or a tumor cell driving its own growth.
The Dolichol-Linked Oligosaccharide Pathway
The 14-sugar glycan that ends up attached to asparagine is not built directly on the protein. It is preassembled on a special lipid carrier called dolichol phosphate, a polyisoprenoid alcohol embedded in the ER membrane. The biosynthetic pathway involves about 12 enzymes acting in sequence:
- UDP-GlcNAc + dolichol-P → dolichol-PP-GlcNAc (catalyzed by DPAGT1, the first committed step)
- Addition of a second GlcNAc → dolichol-PP-GlcNAc2
- Addition of 5 mannose residues (Man5-GlcNAc2) on the cytoplasmic face of the ER membrane
- Flipping of the precursor across the ER membrane to the lumenal face by the flippase RFT1
- Addition of 4 more mannose residues (Man9-GlcNAc2) on the lumenal face
- Addition of 3 glucose residues, completing Glc3Man9GlcNAc2
- Transfer en bloc to a nascent asparagine residue by the OST complex
The OST complex itself is a large multi-subunit assembly (8 subunits in mammals) containing a catalytic STT3 subunit that recognizes the Asn-X-Ser/Thr sequon and catalyzes the glycan transfer. There are two STT3 isoforms in mammals (STT3A and STT3B) with distinct substrate preferences: STT3A acts co-translationally on emerging chains, while STT3B can act post-translationally on skipped sites.
This dolichol-linked oligosaccharide pathway depends on a steady supply of nucleotide sugars (UDP-GlcNAc, GDP-mannose, UDP-glucose), dolichol, and ATP for the various transferase reactions. Defects in any of the enzymes — or in the supply of dolichol or nucleotide sugars — produce specific subtypes of congenital disorders of glycosylation. For example, PMM2-CDG (the most common CDG) is caused by phosphomannomutase 2 deficiency, reducing GDP-mannose supply for steps 3 and 5. The metabolism of all these intermediates is exquisitely sensitive to cellular energy and nutrient status — another reason N-glycosylation, and therefore asparagine demand, scales with cellular activity.
ER and Golgi Processing of N-Glycans
The glycan that ends up on a mature glycoprotein is rarely the original Glc3Man9GlcNAc2. After attachment, the glycan undergoes extensive processing as the protein moves through the secretory pathway:
- In the ER: the three glucose residues are sequentially removed by glucosidases I and II. The single-glucose intermediate is the substrate for the calnexin/calreticulin chaperones (see next section). After folding, the final glucose is removed, and 1-4 mannoses are trimmed by ER mannosidases.
- In the cis Golgi: Golgi mannosidase I removes additional mannoses, producing Man5GlcNAc2.
- In the medial Golgi: GlcNAc transferases add GlcNAc residues, initiating the conversion to complex-type glycans. Additional mannoses are trimmed.
- In the trans Golgi: galactose, fucose, and sialic acid are added by specific glycosyltransferases, producing the final mature glycan.
The result is heterogeneity. A single protein at a single glycosylation site can carry a range of mature glycan structures, from high-mannose (in proteins that move quickly through the pathway) to complex bi-, tri-, and tetra-antennary glycans (in proteins that undergo full Golgi processing). This "glycoprotein microheterogeneity" is a feature, not a bug — different glycan structures direct the protein to different destinations and confer different functional properties.
The clinical relevance of this Golgi processing is illustrated by congenital disorders of glycosylation type II (CDG-II), where defects in Golgi glycosyltransferases produce truncated glycans on otherwise-normal protein backbones. Patients with CDG-II often have severe multisystem disease including immunodeficiency, coagulopathy, and neurodevelopmental delay — reflecting the breadth of glycoproteins whose function is compromised.
Glycoprotein Folding and the Calnexin-Calreticulin Cycle
One of the most elegant uses the cell makes of N-glycans is as a quality-control tag during protein folding. After OST attaches the glycan to a nascent asparagine, glucosidases I and II trim two of the three glucose residues. The mono-glucosylated GlcNAc2Man9Glc1 species is the substrate for the lectin chaperones calnexin (membrane-bound) and calreticulin (soluble in ER lumen), which bind the glycoprotein and recruit ERp57 (a protein disulfide isomerase) to catalyze proper disulfide bond formation.
The clever part: the glycoprotein leaves the chaperone when the last glucose is removed by glucosidase II. If the protein is properly folded, it exits the ER and proceeds through the Golgi. If it is misfolded, it is recognized by UDP-glucose:glycoprotein glucosyltransferase (UGGT), which re-adds a glucose — sending the protein back to calnexin/calreticulin for another folding attempt. This re-glucosylation/de-glucosylation cycle allows multiple attempts at proper folding, with the cell's quality-control machinery monitoring each cycle. Persistent misfolding eventually triggers ER-associated degradation (ERAD), in which the protein is retro-translocated to the cytoplasm and degraded by the proteasome.
This cycle is the central pillar of secretory pathway quality control. It ensures that only properly folded proteins reach the cell surface or are secreted, preventing misfolded proteins from causing downstream damage. When the system is overwhelmed (by mutated proteins, ER stress, or massive secretory demand), the unfolded protein response (UPR) is triggered — activating IRE1, PERK, and ATF6 signaling that reduces global translation, upregulates chaperones, and ultimately can trigger apoptosis if the stress persists.
Several human diseases involve failures of this quality-control system. Z-allele alpha-1 antitrypsin deficiency, the most common cause of inherited liver disease in adults, results from a mutation that causes alpha-1 antitrypsin to misfold and polymerize in hepatocyte ER, causing chronic ER stress, hepatocyte injury, and eventually cirrhosis. The misfolded Z-protein accumulates because the calnexin/calreticulin cycle cannot productively refold it, and the ERAD machinery cannot keep up with the production rate.
Immunoglobulin Folding and the IgG-Fc Glycan
Immunoglobulin G is a particularly important and well-studied glycoprotein, both because of its central role in humoral immunity and because its N-glycans serve as both a structural and functional regulator. IgG carries a conserved N-glycosylation site at Asn297 in the Fc region of each heavy chain, and additional variable glycosylation sites on roughly 20% of Fab regions. The Asn297 glycan is essential for proper Fc folding — deglycosylated IgG has reduced thermal stability and altered Fc receptor binding.
The Asn297 glycan is biantennary, with variable terminal modifications:
- Galactosylation — the presence of zero, one, or two terminal galactose residues defines G0, G1, and G2 IgG glycoforms. The G0 glycoform increases in rheumatoid arthritis, lupus, and other autoimmune conditions, and decreases during pregnancy in healthy women.
- Sialylation — addition of terminal sialic acid increases the half-life of circulating IgG and shifts Fc receptor binding toward inhibitory FcγRIIB, producing an anti-inflammatory effect. The mechanism behind the clinical effect of IVIG in autoimmune conditions involves the sialylated minor fraction of IVIG IgG.
- Core fucosylation — the addition of fucose to the inner GlcNAc reduces antibody-dependent cellular cytotoxicity (ADCC). Removing core fucose (afucosylation) enhances ADCC by 50-100 fold — this is the basis for engineered therapeutic antibodies like obinutuzumab (afucosylated anti-CD20 for CLL).
- Bisecting GlcNAc — addition of a bisecting GlcNAc modulates effector function and antibody-dependent cellular phagocytosis.
This Fc glycan heterogeneity is a tunable parameter of immune function. Plasma cells modulate IgG glycosylation in response to cytokine signals, and chronic inflammation shifts the population toward G0 (less anti-inflammatory) glycoforms. The discovery that IVIG's anti-inflammatory effect depends on sialylated IgG has led to research into Fc glycan engineering as a therapeutic strategy.
Asparagine is the single residue that the entire Fc glycan structure attaches to. Mutate Asn297 and the heavy chain misfolds, antibody assembly fails, and humoral immunity collapses. The role of asparagine in immune defense (covered also on the canonical Asparagine page) ultimately reduces to this: every antibody molecule depends on properly glycosylated asparagines.
Congenital Disorders of Glycosylation (CDG)
The CDG family is the rapidly growing class of inborn errors of glycosylation. First recognized in 1980 by Jaeken, the number of distinct CDG subtypes has grown from 1 to more than 150 over four decades, with new genes continuing to be added. CDGs are divided into two broad groups:
- CDG-I (Type I) — defects in the assembly of the dolichol-linked oligosaccharide precursor or its transfer to protein. The most common is PMM2-CDG (phosphomannomutase 2 deficiency), formerly called CDG-Ia. Patients have multisystem disease including hypotonia, developmental delay, abnormal subcutaneous fat distribution, inverted nipples, and progressive cerebellar atrophy.
- CDG-II (Type II) — defects in the Golgi processing of N-glycans after attachment. Patients have variable phenotypes including immunodeficiency (loss of properly glycosylated immune receptors), coagulopathy (loss of properly glycosylated clotting factors), and developmental delay.
The clinical diagnosis of CDG often starts with the screening test of plasma transferrin isoelectric focusing or mass spectrometry, which reveals abnormal patterns of transferrin glycoforms (transferrin normally has 2 N-glycans, each with 2 sialic acids, totaling 4 sialic acids per transferrin molecule; CDG patients have an abnormal distribution of less-glycosylated species). The specific CDG subtype is then identified by genetic testing or by enzymatic assay of fibroblasts.
Treatment is largely supportive, though a few CDG subtypes have specific therapies: MPI-CDG (mannose phosphate isomerase deficiency) responds dramatically to oral mannose supplementation, and SLC35C1-CDG (GDP-fucose transporter deficiency) responds to oral fucose. Most other CDGs lack specific therapy and are managed with supportive care for the affected organ systems.
The CDG family is collectively quite rare (incidence in aggregate is probably 1 in 50,000 to 1 in 100,000), but is increasingly recognized as a cause of unexplained multisystem disease, particularly in infants and children. Whole-exome sequencing has been instrumental in identifying the rapidly expanding list of new CDG genes.
Secreted Proteins, Membrane Proteins, and Cell-Surface Receptors
The functional reach of N-glycosylation includes essentially every protein the cell exports or displays on its surface. Examples by category:
- Plasma proteins — albumin (no N-glycans, the exception), transferrin (2 sites), ceruloplasmin (4 sites), alpha-1 antitrypsin (3 sites), all complement components, IgG, IgA, IgM. Liver albumin synthesis bypasses N-glycosylation, but virtually every other liver-secreted plasma protein depends on it.
- Hormones — erythropoietin (3 N-glycans, critical for half-life), follicle-stimulating hormone (2 on alpha, 2 on beta), thyroid-stimulating hormone (2 on alpha, 1 on beta), human chorionic gonadotropin (2 on alpha, 4 on beta), luteinizing hormone, gonadotropins. Hormone glycosylation regulates not only half-life but also receptor binding kinetics.
- Cell-surface receptors — insulin receptor, IGF-1 receptor, EGF receptor, all growth factor receptors, all cytokine receptors, MHC class I and class II, T-cell receptor, B-cell receptor. Receptor glycosylation modulates ligand affinity and receptor trafficking.
- Adhesion molecules — selectins, integrins, cadherins, immunoglobulin-superfamily adhesion proteins. Cell-cell and cell-matrix adhesion is heavily regulated by N-glycan structures, particularly the terminal sialic acid and sulfate groups.
- Coagulation factors — factor V, VII, VIII, IX, X, XI, XII, von Willebrand factor, fibrinogen. Coagulopathy is one of the cardinal features of severe CDGs because so many clotting factors depend on N-glycosylation.
- Lysosomal enzymes — targeting to lysosomes depends on the mannose-6-phosphate tag added to specific N-glycans of soluble lysosomal proteins.
The collective demand for asparagine in N-glycosylation across all these proteins is substantial. Most cells turn over their entire glycoprotein complement on a timescale of hours to days. Plasma cells secreting antibodies can produce up to several thousand IgG molecules per second — each requiring multiple glycosylated asparagines — making them among the most asparagine-demanding cell types in the body.
Other Post-Translational Roles of Asparagine
Beyond N-glycosylation, asparagine residues serve several other specialized post-translational roles:
- Asparagine-hydroxylation — the hypoxia-inducible factor (HIF) family of transcription factors is regulated in part by asparagine hydroxylation. Under normoxic conditions, factor inhibiting HIF (FIH) hydroxylates a specific asparagine in HIF-1-alpha's C-terminal transactivation domain, preventing recruitment of the p300/CBP co-activator and suppressing HIF target gene expression. Under hypoxia, the hydroxylation is blocked, HIF becomes transcriptionally active, and the hypoxic response (including erythropoietin upregulation) ensues.
- Asparagine-isomerization — asparagine residues are subject to spontaneous deamidation and isomerization to isoaspartate, particularly when followed by glycine in the sequence. This is the molecular basis of the "asparagine clock" for protein aging (see next section).
- Beta-aspartyl shift — in some proteolytic cleavage reactions, asparagine can undergo a cyclic imide intermediate that rearranges the peptide bond from alpha to beta linkage.
- N-terminal asparagine — when asparagine is the N-terminal residue of a protein, it is a destabilizing residue under the N-end rule, marking the protein for ubiquitination and proteasomal degradation. N-terminal asparagine is converted to aspartate by N-terminal asparagine amidohydrolase (NTAN1), and N-terminal aspartate is then conjugated to arginine by ATE1 (arginyltransferase), creating the N-degron recognized by Ubr1 E3 ligases.
Asparagine Deamidation and the Molecular Clock of Aging
One of the most intriguing properties of asparagine is its spontaneous chemical instability in proteins. Under physiologic conditions, the asparagine side-chain amide undergoes slow hydrolysis — the amide nitrogen is replaced by a hydroxyl, converting asparagine to aspartate (or via a succinimide intermediate, to iso-aspartate). The half-life of this deamidation depends critically on the flanking sequence:
- Asn-Gly is the fastest, with a deamidation half-life of approximately 1-2 days at physiologic pH and temperature
- Asn-Ala, Asn-Ser have intermediate half-lives of weeks to months
- Asn-Pro, Asn-Val are slow, with half-lives of years
This sequence-dependent deamidation has been proposed by Arthur Robinson as a molecular clock for protein aging. Long-lived proteins (lens crystallins, articular cartilage collagen, elastin in arteries) progressively accumulate deamidated asparagine residues over decades, and this accumulation correlates with age-related protein dysfunction. The accumulation of deamidated/isomerized residues has been implicated in the brown coloration of aged eye lens crystallins (a contributor to cataract formation), in the loss of elasticity in aging arteries, and in the conformational changes of amyloid-beta peptide that may contribute to Alzheimer's pathology.
The cell has a partial repair mechanism in protein-L-isoaspartyl methyltransferase (PIMT, encoded by PCMT1), which methylates the alpha-carboxylate of isoaspartate residues, initiating a rearrangement that can restore some fraction back to native aspartate (with the iso-Asp/Asp ratio shifted slightly toward Asp). Mice with PIMT knockout develop fatal seizures by 30-60 days of age — consistent with the brain's particular sensitivity to accumulated isoaspartate damage. Human PIMT polymorphisms have been associated with neurodegenerative disease susceptibility, though this remains an active research area.
Cautions and Clinical Considerations
- Suspect CDG in any infant with unexplained multisystem disease — particularly with hypotonia, abnormal fat distribution, inverted nipples, coagulopathy, or unexplained transaminitis. Plasma transferrin isoelectric focusing is the standard screening test.
- MPI-CDG responds to oral mannose — this is one of the few treatable inborn errors of glycosylation. Diagnosis is critical because untreated MPI-CDG produces protein-losing enteropathy that responds dramatically to mannose supplementation.
- Pregnancy-induced changes in IgG glycosylation — the increase in galactosylated and sialylated IgG during pregnancy is one mechanism behind the spontaneous remission of rheumatoid arthritis often observed during pregnancy. Disease typically flares postpartum as glycosylation patterns revert.
- Tunicamycin (an antibiotic that inhibits N-glycosylation by blocking DPAGT1) is a research tool, not a clinical drug; its use illustrates the cellular catastrophe of complete N-glycosylation failure, which causes massive ER stress and apoptosis.
- Therapeutic antibody engineering — modern biotechnology routinely manipulates antibody glycosylation to enhance ADCC (afucosylation as in obinutuzumab), reduce immunogenicity (humanization of glycans), or extend half-life. Patients receiving these drugs should be aware that the glycan composition is part of the drug's mechanism.
- Aged proteins and disease — the asparagine deamidation clock means certain long-lived proteins (lens crystallins, articular collagen) accumulate molecular damage with age that no nutritional intervention can reverse. Prevention strategies (UV protection for the lens, joint mechanics for cartilage) are more effective than corrective interventions.
- Asparagine supplementation does not bypass CDG defects — the bottleneck in N-glycosylation defects is the enzyme machinery, not the supply of asparagine residues in nascent proteins. Dietary asparagine cannot rescue a defective OST complex or a defective dolichol pathway.
Key Research Papers
- Helenius A, Aebi M (2004). Roles of N-linked glycans in the endoplasmic reticulum. Annual Review of Biochemistry, 73:1019-1049. — PubMed: Helenius Aebi review
- Kornfeld R, Kornfeld S (1985). Assembly of asparagine-linked oligosaccharides. Annual Review of Biochemistry, 54:631-664. — PubMed: Kornfeld N-glycan assembly
- Aebi M (2013). N-linked protein glycosylation in the ER. Biochimica et Biophysica Acta, 1833(11):2430-2437. — PubMed: Aebi N-glycosylation
- Jaeken J, Matthijs G (2007). Congenital disorders of glycosylation: a rapidly expanding disease family. Annual Review of Genomics and Human Genetics, 8:261-278. — PubMed: Jaeken CDG review
- Freeze HH, Schachter H, Kinoshita T (2017). Genetic Disorders of Glycosylation. In: Essentials of Glycobiology. — PubMed: Freeze CDG essentials
- Anelli T, Sitia R (2008). Protein quality control in the early secretory pathway. EMBO Journal, 27(2):315-327. — PubMed: Anelli ERAD quality control
- Hebert DN, Molinari M (2007). In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiological Reviews, 87(4):1377-1408. — PubMed: ER protein folding review
- Anthony RM, Wermeling F, Karlsson MC, Ravetch JV (2008). Identification of a receptor required for the anti-inflammatory activity of IVIG. PNAS, 105(50):19571-19578. — PubMed: Anthony IVIG receptor
- Robinson NE, Robinson AB (2001). Molecular clocks. PNAS, 98(3):944-949. — PubMed: Robinson molecular clock
- Reissner KJ, Aswad DW (2003). Deamidation and isoaspartate formation in proteins: unwanted alterations or surreptitious signals? Cellular and Molecular Life Sciences, 60(7):1281-1295. — PubMed: Reissner deamidation review
- Kim E et al. (1997). Deficiency of a protein-repair enzyme results in the accumulation of altered proteins, retardation of growth, and fatal seizures in mice. PNAS, 94(12):6132-6137. — PubMed: PIMT knockout mice
- Bohne-Lang A, von der Lieth CW (2005). GlyProt: in silico glycosylation of proteins. Nucleic Acids Research, 33(W):W214-W219. — PubMed: Glycosylation site prediction
- Reily C et al. (2019). Glycosylation in health and disease. Nature Reviews Nephrology, 15(6):346-366. — PubMed: Reily glycosylation review
PubMed Topic Searches
- PubMed: N-glycosylation sequon
- PubMed: OST mechanism
- PubMed: CDG treatment
- PubMed: IgG Fc glycosylation
- PubMed: Asparagine deamidation aging
Connections
- Asparagine Benefits Hub
- Asparagine Overview
- Nervous System Function
- Ammonia Detoxification
- Asparaginase Therapy
- Glutamine
- Serine (Sequon Partner)
- Threonine (Sequon Partner)
- Aspartic Acid
- Glutamic Acid
- Manganese (Glycosyltransferase Cofactor)
- Magnesium
- Liver Disease (Alpha-1 Antitrypsin)
- Anemia (Erythropoietin Glycosylation)
- All Amino Acids