L-Asparaginase Therapy for Pediatric ALL
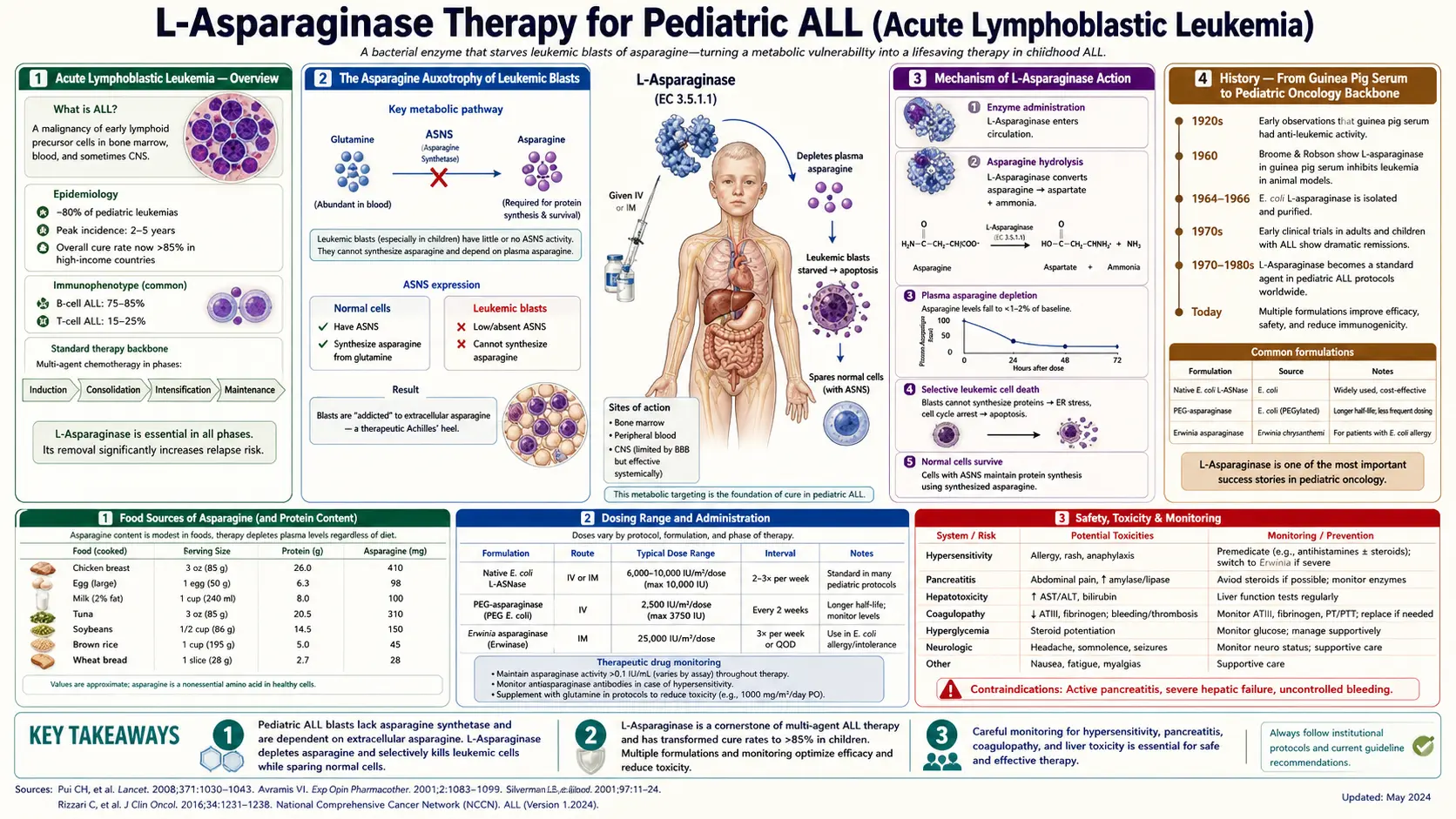
Acute lymphoblastic leukemia (ALL) of childhood is the prototype curable cancer of the modern era — five-year overall survival in pediatric ALL has risen from roughly 10% in the 1960s to better than 90% today. No single drug has played a larger role in that transformation than L-asparaginase. The drug exploits a metabolic vulnerability so specific to leukemic blasts that it borders on poetic: virtually every healthy tissue can make its own asparagine on demand, but the lymphoid progenitor cells that become ALL silence the asparagine synthetase (ASNS) gene during their normal developmental program, becoming "asparagine auxotrophs" that depend on the plasma pool to survive. Inject the bacterial enzyme L-asparaginase (which hydrolyzes plasma asparagine to aspartate and ammonia, depleting plasma asparagine to undetectable levels for days at a time), and the leukemic blasts cannot make protein, cannot grow, and die by apoptosis. The pediatric ALL backbone today still includes 30+ doses of asparaginase across induction, consolidation, and intensification — first as native E. coli asparaginase (Elspar, now largely retired), then as PEGylated E. coli asparaginase (Oncaspar, Asparlas), and as Erwinia chrysanthemi asparaginase (Erwinaze, Rylaze) for patients who develop hypersensitivity to the E. coli products. This page walks through the biology, history, formulations, toxicities, and management of the most metabolic of all cancer drugs.
Table of Contents
- Acute Lymphoblastic Leukemia — Overview
- The Asparagine Auxotrophy of Leukemic Blasts
- Mechanism of L-Asparaginase Action
- History — From Guinea Pig Serum to Pediatric Oncology Backbone
- Modern Asparaginase Formulations
- Hypersensitivity Reactions and the Erwinia Switch
- Non-Allergic Toxicities — Pancreatitis, Coagulopathy, Hepatotoxicity
- Therapeutic Drug Monitoring of Asparaginase Activity
- Resistance Mechanisms and ASNS Upregulation
- Adult ALL and Pediatric-Inspired Regimens
- Cautions and Clinical Considerations
- Key Research Papers
- Connections
- Featured Videos
Acute Lymphoblastic Leukemia — Overview
Acute lymphoblastic leukemia is the most common childhood cancer, accounting for approximately 25% of all pediatric malignancies and approximately 75% of childhood leukemias. Annual U.S. incidence is approximately 3,000 to 4,000 new cases of pediatric ALL, with a peak incidence at age 2-5 years. The disease arises from malignant transformation of lymphoid progenitor cells in the bone marrow, with subsequent infiltration of marrow, lymph nodes, central nervous system, and other tissues.
Pediatric ALL is biologically divided into B-cell precursor ALL (B-ALL, approximately 85% of cases) and T-cell ALL (T-ALL, approximately 15%), each with characteristic genetic subtypes that drive risk stratification. The most common cytogenetic abnormalities in B-ALL include high hyperdiploidy (47-50 chromosomes, favorable prognosis), the ETV6-RUNX1 (formerly TEL-AML1) translocation (favorable), and the BCR-ABL1 (Philadelphia chromosome) translocation (historically poor prognosis, now markedly improved by tyrosine kinase inhibitors). T-ALL carries activating mutations in NOTCH1 in approximately 60% of cases.
Modern ALL therapy is divided into three phases:
- Induction (4-5 weeks) — multi-agent chemotherapy aiming for morphologic remission. Standard backbone includes vincristine, a glucocorticoid (dexamethasone or prednisone), anthracycline (daunorubicin), and asparaginase, with intrathecal methotrexate for CNS prophylaxis.
- Consolidation/Intensification (several months) — high-dose methotrexate, additional asparaginase doses, mercaptopurine, cyclophosphamide, cytarabine, and CNS-directed therapy.
- Maintenance (2-3 years) — daily oral mercaptopurine plus weekly oral methotrexate, with periodic pulses of vincristine and glucocorticoid. Most modern protocols include additional asparaginase pulses in this phase as well.
The total duration of pediatric ALL therapy is 2 to 3 years from diagnosis, with cumulative asparaginase doses ranging from 8-9 (older regimens) to 30+ (modern intensive regimens) across the entire treatment course.
The Asparagine Auxotrophy of Leukemic Blasts
The biological basis for L-asparaginase's remarkable activity in ALL lies in a single distinctive feature of leukemic lymphoblasts: they fail to express asparagine synthetase (ASNS), the enzyme that converts aspartate and glutamine into asparagine. Healthy cells throughout the body express ASNS in proportion to their proliferative demand, allowing them to generate asparagine intracellularly when plasma supply is reduced. Most ALL blasts lack this ability and depend on plasma asparagine.
This loss of ASNS expression appears to be a developmental remnant of normal lymphoid biology. During normal lymphoid maturation, ASNS expression varies across developmental stages. Naive lymphocytes in resting peripheral blood have relatively low ASNS expression but high baseline asparagine levels supplied by plasma. The leukemic transformation seems to lock the lymphoblast in a state of low ASNS expression while simultaneously driving very high proliferation rates — producing a cell with high asparagine demand and no ability to make its own.
When plasma asparagine is depleted (by L-asparaginase therapy), normal cells throughout the body activate the integrated stress response (GCN2 → eIF2-alpha-P → ATF4 → ASNS transcription), upregulate ASNS within hours, and begin making their own asparagine to compensate. Most leukemic blasts cannot do this — the ASNS gene is epigenetically silenced or transcriptionally repressed by leukemia-specific transcription factors — and so they cannot escape the asparagine depletion. They halt protein synthesis, fail to progress through the cell cycle, and undergo apoptosis through the integrated stress response's pro-apoptotic arm (CHOP/GADD153 activation).
This auxotrophic vulnerability is not unique to ALL among hematologic malignancies but is most pronounced in lymphoblastic disease. NK/T-cell lymphomas and some myeloid leukemias also show asparagine dependence, but the degree of clinical response to L-asparaginase has been most consistent and dramatic in pediatric B-precursor and T-cell ALL.
Mechanism of L-Asparaginase Action
L-asparaginase is the bacterial enzyme (most commonly purified from Escherichia coli or Erwinia chrysanthemi) that catalyzes the hydrolysis of L-asparagine to L-aspartate and ammonia:
L-asparagine + H2O → L-aspartate + NH3
The mammalian enzyme that catalyzes the reverse reaction (asparagine synthetase, ASNS) is described in detail on our Ammonia Detoxification page. Bacterial L-asparaginase is unrelated structurally to mammalian ASNS and works in the opposite direction.
Pharmacologically, intravenous L-asparaginase rapidly depletes plasma asparagine. A typical dose reduces plasma asparagine from 40-80 µmol/L to undetectable levels (less than 0.5 µmol/L) within hours, and depletion persists for days (native E. coli asparaginase) to weeks (PEG-asparaginase) depending on the formulation's pharmacokinetics. Native E. coli asparaginase has a half-life of approximately 1 day; PEGylated asparaginase has a half-life of approximately 5-7 days; Erwinia asparaginase has a half-life of approximately half a day.
Importantly, most L-asparaginase formulations also have low-level glutaminase activity — they hydrolyze glutamine to glutamate and ammonia at perhaps 2-10% of the asparaginase activity. This dual activity contributes both to the drug's efficacy (depleting glutamine, which leukemic blasts use as an asparagine precursor) and to several of its toxicities (the glutamine depletion contributes to coagulopathy, hepatotoxicity, and CNS effects). Erwinia asparaginase has lower glutaminase activity than E. coli asparaginase, which is one reason it is generally better tolerated.
The complete mechanism summary:
- IV bolus or IM injection of L-asparaginase
- Rapid hydrolysis of plasma asparagine to aspartate + ammonia
- Plasma asparagine drops to undetectable levels within hours
- Slower, secondary depletion of plasma glutamine through low-level glutaminase activity
- Leukemic blasts (which cannot upregulate ASNS) lose asparagine supply for protein synthesis
- Integrated stress response activation in blasts → halt of protein synthesis → pro-apoptotic CHOP induction → apoptosis
- Normal tissues, which upregulate ASNS, continue making asparagine and survive
History — From Guinea Pig Serum to Pediatric Oncology Backbone
The discovery of L-asparaginase's antileukemic activity is one of the great serendipitous stories of mid-20th-century oncology. In 1953, John Kidd, working at the Sloan-Kettering Institute, observed that guinea pig serum — but not serum from other mammals — produced regression of certain transplanted murine lymphomas. He could not identify the active component but published the observation as a curious phenomenon.
For nearly a decade, the "guinea pig factor" remained unexplained. In 1961, John Broome at the University of Maryland identified it: the antitumor activity of guinea pig serum was due to its unusually high L-asparaginase activity (guinea pigs are the rare mammal with substantial circulating asparaginase). Broome demonstrated that purified bacterial L-asparaginase (initially from E. coli) could reproduce the antitumor effect, and that the mechanism was depletion of plasma asparagine.
The first clinical trials in children with refractory ALL took place in the mid-1960s, with dramatic results — complete remission rates of 30-60% as a single agent in heavily pretreated children. By 1968, asparaginase was being incorporated into combination regimens, and in the 1970s it became a standard component of induction therapy. The drug was FDA-approved as Elspar (native E. coli asparaginase) in 1978.
The next major advance was PEGylation. Native E. coli asparaginase is highly immunogenic — up to 40% of patients develop anti-asparaginase antibodies, with some experiencing severe hypersensitivity reactions and others showing "silent inactivation" (antibody-mediated clearance without overt allergy, leading to subtherapeutic drug levels). Covalently attaching polyethylene glycol chains to the enzyme reduces immunogenicity, extends half-life, and reduces the dosing frequency. PEG-asparaginase (Oncaspar, FDA-approved in 1994) became the preferred first-line asparaginase preparation in most pediatric regimens by the 2000s.
For patients who develop hypersensitivity to E. coli-derived asparaginases (native or PEG), the alternative is Erwinia chrysanthemi asparaginase, immunologically distinct from E. coli enzyme. Erwinia asparaginase (Erwinaze, FDA-approved 2011; replaced by Rylaze recombinant Erwinia in 2021 due to manufacturing supply issues with the native product) has lower glutaminase activity and is generally well-tolerated even in patients who have failed E. coli products.
The most recent formulation is calaspargase pegol (Asparlas, FDA-approved 2018), a longer-acting PEG-asparaginase that allows reduced dosing frequency (every 21 days instead of every 14 days), particularly in the consolidation/maintenance phases.
Modern Asparaginase Formulations
Four asparaginase preparations are in active clinical use as of the mid-2020s:
| Drug | Source | Half-Life | Notes |
|---|---|---|---|
| Native E. coli asparaginase (Elspar) | Wild-type E. coli | ~1 day | Largely retired; high immunogenicity |
| PEG-asparaginase (Oncaspar) | PEGylated E. coli | ~5-7 days | Standard first-line; reduced immunogenicity |
| Calaspargase pegol (Asparlas) | PEGylated E. coli, succinimidyl carbonate linker | ~10-16 days | Longer-acting; q21-day dosing |
| Erwinia asparaginase (Rylaze, formerly Erwinaze) | Recombinant Erwinia chrysanthemi | ~0.5 days | For E. coli hypersensitivity; lower glutaminase activity |
Choice of agent depends on the patient's allergic history, the treatment protocol, and the phase of therapy. PEG-asparaginase is the standard first-line agent in most modern pediatric ALL protocols (COG, DFCI, Berlin-Frankfurt-Münster). Calaspargase pegol is increasingly used in the maintenance phase to reduce visit frequency. Erwinia asparaginase is reserved for patients with documented hypersensitivity or silent inactivation of E. coli-derived asparaginases.
Dosing varies by protocol but typical examples for pediatric B-ALL:
- PEG-asparaginase: 2,500 IU/m2 IM or IV, single dose per cycle, q14 days during intensification phase
- Calaspargase pegol: 2,500 IU/m2 IV, q21 days
- Erwinia (Rylaze): 25 mg/m2 IM Mon/Wed/Fri for 6 doses (~2 weeks) to replace one dose of PEG-asparaginase
Hypersensitivity Reactions and the Erwinia Switch
Hypersensitivity to L-asparaginase is the most frequent reason to switch formulations. Native E. coli asparaginase produced clinical hypersensitivity in 30-45% of patients across the course of therapy; PEG-asparaginase reduces this rate to approximately 5-25% depending on the regimen and the cumulative number of doses.
Clinical manifestations range from mild (transient urticaria, isolated wheezing) to severe (laryngeal edema, hypotension, anaphylaxis requiring epinephrine). Reactions typically occur during or shortly after infusion. Premedication with antihistamines and glucocorticoids does not reliably prevent severe reactions and is not standard practice in most modern protocols, because it can mask the diagnostic features of a hypersensitivity reaction without preventing the underlying immunologic process.
A particular concern is "silent inactivation" or "subclinical hypersensitivity" — patients who do not have overt allergic symptoms but who have developed neutralizing antibodies that clear the drug rapidly. These patients receive subtherapeutic asparagine depletion (plasma asparagine remains elevated despite asparaginase dosing), reducing the antileukemic effect without producing any clinical warning sign. Modern protocols therefore include therapeutic drug monitoring of asparaginase activity at specific timepoints (see next section), with a switch to Erwinia preparation for any patient showing inadequate activity even without overt allergy.
The standard switching algorithm:
- Initial therapy with PEG-asparaginase
- If clinical hypersensitivity (grade 2 or higher) develops during PEG-asparaginase → switch to Erwinia asparaginase for remaining doses
- If asparaginase activity monitoring shows subtherapeutic levels (silent inactivation) → switch to Erwinia
- If hypersensitivity develops to both E. coli and Erwinia preparations → consider re-challenge with PEG under desensitization protocol, or omit asparaginase if alternative intensification is feasible
The Berlin-Frankfurt-Münster (BFM) group's data show that patients who complete the planned asparaginase dose intensity (whether through PEG or Erwinia substitution) have superior event-free survival compared to patients with incomplete asparaginase exposure. This drove the implementation of routine asparaginase activity monitoring across most pediatric oncology cooperative groups.
Non-Allergic Toxicities — Pancreatitis, Coagulopathy, Hepatotoxicity
L-asparaginase causes a distinctive constellation of non-allergic toxicities that reflect the breadth of physiologic processes that depend on plasma asparagine and glutamine. The most consequential are:
- Acute pancreatitis — occurs in approximately 5-10% of patients receiving PEG-asparaginase, often severe enough to require ICU admission. Some cases progress to pseudocyst formation or chronic pancreatitis. The mechanism involves direct injury to pancreatic acinar cells, which have very high asparagine demand for digestive enzyme production. Asparaginase therapy is permanently discontinued after a single episode of severe pancreatitis — rechallenge has unacceptably high rates of recurrent pancreatitis.
- Coagulopathy and thrombosis — the liver synthesizes virtually all clotting factors and clotting inhibitors, and many of these are heavily glycosylated proteins requiring asparagine. Asparaginase depletes asparagine and glutamine, impairing hepatic synthesis of antithrombin (the most common deficit), protein C, protein S, fibrinogen, and factors II, VII, IX, X. The clinical picture is paradoxically thrombotic rather than bleeding, because antithrombin deficiency dominates. Cerebral venous sinus thrombosis (CVST) is a feared complication, occurring in 1-3% of patients with PEG-asparaginase. Prophylactic antithrombin replacement and/or anticoagulation in high-risk patients (older adolescents, obese patients) is increasingly common.
- Hepatotoxicity — elevated transaminases occur in 20-50% of patients, ranging from mild grade 1 elevations to severe hepatitis with hyperbilirubinemia. Fatty liver is common. Most cases resolve with dose delay; severe hepatitis may require permanent discontinuation. Steatosis can persist for months after therapy.
- Hyperglycemia and pancreatic islet dysfunction — the pancreatic islet cells, like acinar cells, are sensitive to asparagine depletion. Insulin secretion may be impaired transiently, producing hyperglycemia. Combined with concomitant glucocorticoid therapy, this can produce overt diabetes mellitus during treatment, sometimes requiring insulin therapy.
- Hypertriglyceridemia — serum triglycerides commonly rise dramatically (sometimes > 1,000 mg/dL) during asparaginase therapy. This typically reflects impaired hepatic synthesis of apolipoproteins and altered lipoprotein lipase activity. Most cases resolve spontaneously; severe cases may require fenofibrate or temporary asparaginase dose adjustment.
- Neurotoxicity — cerebral venous sinus thrombosis (mentioned above) is the most serious CNS complication. Posterior reversible encephalopathy syndrome (PRES) has also been described. Direct asparaginase neurotoxicity is rare because of the CNS's buffering by local ASNS-mediated asparagine synthesis (see Nervous System Function page).
Management of these toxicities is largely supportive (dose delay for moderate toxicity, dose reduction or discontinuation for severe toxicity, antithrombin replacement for CVST risk). The challenge is that omitting asparaginase entirely is associated with worse leukemia outcomes, so the oncology team navigates a balance between toxicity management and dose intensity preservation.
Therapeutic Drug Monitoring of Asparaginase Activity
Modern asparaginase use includes routine measurement of serum asparaginase activity at protocol-specified timepoints (typically days 7, 14, and 21 after each PEG-asparaginase dose). This monitoring serves multiple purposes:
- Detection of silent inactivation (subclinical antibody-mediated clearance)
- Confirmation that protocol-targeted activity is being achieved
- Guidance for switching to Erwinia formulation if activity is inadequate
- Pharmacokinetic data to refine dosing in special populations (obesity, organ dysfunction)
The threshold for adequate activity is typically > 0.1 IU/mL at the trough timepoint (day 7 for PEG-asparaginase, day 4 for native E. coli). Activity below this threshold indicates inadequate asparagine depletion and prompts a switch to Erwinia asparaginase for subsequent doses.
Serum asparagine concentration could in principle be measured directly, but the assay is technically demanding (samples must be processed immediately to prevent in vitro asparaginase activity continuing to deplete asparagine in the sample tube). Most centers therefore use asparaginase activity rather than direct asparagine measurement.
Resistance Mechanisms and ASNS Upregulation
The most important resistance mechanism to L-asparaginase is upregulation of ASNS expression in the leukemic blasts. Subclones that retain or regain the ability to express ASNS can synthesize asparagine intracellularly even when plasma asparagine is depleted, escaping the drug's effect. ASNS upregulation can occur through several mechanisms:
- Activation of the integrated stress response — the same pathway healthy cells use (GCN2 → ATF4 → ASNS promoter activation). Leukemic blasts that retain functional GCN2 signaling can sometimes activate this pathway.
- Epigenetic reactivation of ASNS — the ASNS promoter is silenced by DNA methylation in some ALL subtypes; treatment-related selection can favor blasts with hypomethylated ASNS promoters.
- WNT pathway activation — WNT signaling has been shown to drive ASNS expression in ALL cell lines; WNT-active subclones are relatively resistant to asparaginase.
- NRF2 pathway activation — the oxidative stress response transcription factor NRF2 (NFE2L2) drives ASNS expression as part of the broader cellular stress response.
Pre-treatment ASNS expression is a recognized prognostic marker in some pediatric ALL series, with high-ASNS-expressing leukemias having worse outcomes despite intensive asparaginase therapy. This has driven interest in second-generation asparagine-depleting therapies, including engineered human enzymes (to reduce immunogenicity), enzyme variants with enhanced glutaminase activity (to deplete glutamine, the asparagine precursor, more thoroughly), and combination strategies pairing asparaginase with ASNS-targeting agents.
Pediatric T-ALL is particularly interesting because some T-ALL subtypes are exquisitely asparaginase-sensitive (driving the > 80% cure rates in modern protocols) while others — particularly the early T-cell precursor (ETP) subtype — show high baseline ASNS expression and inferior asparaginase responsiveness. ETP-ALL accordingly has historically poorer outcomes than other T-ALL subtypes.
Adult ALL and Pediatric-Inspired Regimens
Adult ALL has historically had much worse outcomes than pediatric ALL — 5-year survival in adults aged 18-39 was approximately 30-40% in older series, compared to > 85% in children. A major insight of the 2000s was that adolescent and young adult (AYA) patients treated on pediatric protocols had superior outcomes compared to those treated on traditional adult regimens, despite the same disease biology. The difference appears to be the more intensive use of asparaginase, glucocorticoid, vincristine, and methotrexate in pediatric protocols.
This insight led to the widespread adoption of "pediatric-inspired regimens" for AYA ALL patients (typically defined as age 18-39 or 18-49), with PEG-asparaginase incorporated at doses similar to pediatric protocols. Examples include CALGB 10403 (an adapted pediatric COG regimen), the DFCI ALL Consortium protocols, the French GRAALL regimen, and the German GMALL/AYA. Outcomes in AYA patients on pediatric-inspired protocols approach those of children with similar leukemia biology.
Toxicity is the limiting factor in older adults — patients over 40 tolerate the asparaginase-rich intensification phases poorly, with high rates of pancreatitis, hepatotoxicity, and thrombosis. Various dose-modified protocols and elderly-specific regimens have been developed for patients over 60, often substituting different intensification components for asparaginase.
Asparaginase is also used in some non-ALL hematologic malignancies, particularly extranodal NK/T-cell lymphoma (where the SMILE and AspaMetDex regimens demonstrate dramatic activity), but its primary indication remains pediatric and AYA ALL.
Cautions and Clinical Considerations
- Pancreatitis is a permanent contraindication to further asparaginase after a single severe episode. Rechallenge has unacceptably high rates of recurrent severe pancreatitis. Patients should be counseled about this risk.
- Cerebral venous sinus thrombosis presents with new severe headache, focal neurologic deficit, or seizure during or shortly after asparaginase therapy. Urgent MRI/MRV is indicated for any concerning symptoms. Prophylactic antithrombin replacement is increasingly common in high-risk patients.
- Hypertriglyceridemia > 1,000 mg/dL is associated with pancreatitis risk; treat with fenofibrate or omega-3 supplementation and consider dose delay.
- Hyperglycemia requires monitoring of fasting glucose; many patients require insulin during the intensification phase, often resolving after treatment completion.
- Pregnancy: asparaginase is teratogenic in animal models. Patients of reproductive potential should use effective contraception during therapy and for at least 3 months after the last dose.
- Live vaccines are contraindicated during asparaginase therapy due to T-cell immunosuppression. Live vaccinations (varicella, MMR) should be deferred until at least 6 months after completion of all chemotherapy.
- Diet during therapy — conventional dietary asparagine intake does not meaningfully blunt the plasma depletion produced by asparaginase, because intestinal asparaginase activity in the leukemic patient continues to clear ingested asparagine. Patients can eat normal diets without compromising drug efficacy.
- Asparagine supplementation would in principle counteract the drug; patients should avoid amino-acid supplements containing free asparagine during asparaginase therapy.
- Hypersensitivity desensitization protocols exist but are reserved for patients with no other asparaginase option (failed both E. coli and Erwinia), as the procedure is complex and carries its own risks.
Key Research Papers
- Kidd JG (1953). Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. Journal of Experimental Medicine, 98(6):565-582. — PubMed: Kidd 1953
- Broome JD (1961). Evidence that the L-asparaginase activity of guinea pig serum is responsible for its antilymphoma effects. Nature, 191:1114-1115. — PubMed: Broome 1961
- Pieters R et al. (2011). L-asparaginase treatment in acute lymphoblastic leukemia: a focus on Erwinia asparaginase. Cancer, 117(2):238-249. — PubMed: Pieters asparaginase review
- Hijiya N, van der Sluis IM (2016). Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leukemia & Lymphoma, 57(4):748-757. — PubMed: Asparaginase toxicity review
- Stock W et al. (2019). A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: results of CALGB 10403. Blood, 133(14):1548-1559. — PubMed: CALGB 10403
- Silverman LB et al. (2010). Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed ALL. Leukemia, 24(2):320-334. — PubMed: DFCI ALL Consortium
- Avramis VI, Panosyan EH (2005). Pharmacokinetic/pharmacodynamic relationships of asparaginase formulations: the past, the present and recommendations for the future. Clinical Pharmacokinetics, 44(4):367-393. — PubMed: Asparaginase PK/PD
- Tong WH et al. (2014). A prospective study on drug monitoring of PEGasparaginase and Erwinia asparaginase and asparaginase antibodies in pediatric acute lymphoblastic leukemia. Blood, 123(13):2026-2033. — PubMed: Asparaginase monitoring
- Aslanian AM, Kilberg MS (2001). Multiple adaptive mechanisms to chronic L-asparaginase exposure in leukemic cells. Biochemical Journal, 357(Pt 2):397-405. — PubMed: Asparaginase resistance mechanisms
- Holleman A et al. (2004). Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. NEJM, 351(6):533-542. — PubMed: Holleman gene expression ALL
- Egler RA et al. (2016). L-asparaginase in the treatment of patients with acute lymphoblastic leukemia. Journal of Pharmacology & Pharmacotherapeutics, 7(2):62-71. — PubMed: Egler asparaginase review
- Heo YA, Syed YY, Keam SJ (2019). Pegaspargase: a review in acute lymphoblastic leukaemia. Drugs, 79(7):767-777. — PubMed: Pegaspargase review
- Maese L et al. (2021). Recombinant Erwinia asparaginase (JZP458) in acute lymphoblastic leukemia: results from the phase 2/3 AALL1931 study. Blood, 139(16):2387-2395. — PubMed: Rylaze AALL1931
PubMed Topic Searches
- PubMed: L-asparaginase pediatric ALL
- PubMed: PEG-asparaginase Oncaspar
- PubMed: Erwinia asparaginase hypersensitivity
- PubMed: Asparaginase pancreatitis/coagulopathy
- PubMed: ASNS expression ALL resistance
Connections
- Asparagine Benefits Hub
- Asparagine Overview
- Nervous System Function
- Ammonia Detoxification
- Protein Synthesis & Glycosylation
- Cancer
- Anemia
- Glutamine (Asparagine Precursor)
- Aspartic Acid
- Glutamic Acid
- Liver Disease
- Pancreatitis
- Diabetes / Hyperglycemia
- Thrombosis
- All Amino Acids