Asparagine for Nervous System Function
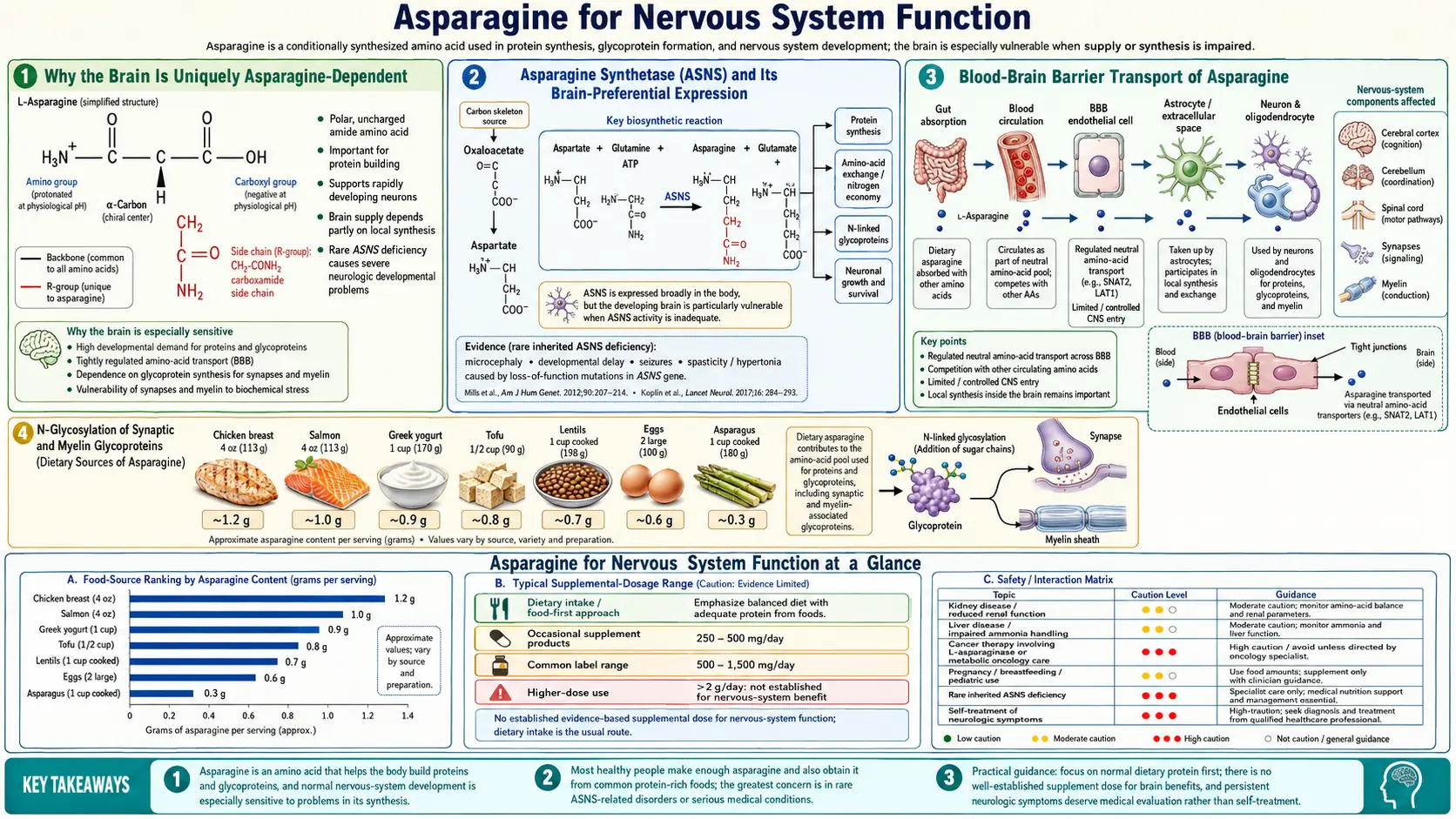
Among the twenty proteinogenic amino acids, asparagine occupies a singular place in the human brain. Although classified as "non-essential" because the body can synthesize it from aspartate and glutamine via asparagine synthetase (ASNS), the central nervous system depends on this local biosynthesis far more than any other organ. Plasma asparagine crosses the blood-brain barrier slowly, so neurons must make most of their own — and when the ASNS gene is mutated, the consequences fall almost entirely on the brain. Asparagine synthetase deficiency (ASNSD), first described in molecular detail by Ruzzo and colleagues in Neuron in 2013, presents at birth with congenital microcephaly, cortical atrophy, intractable seizures, and severe global developmental delay — a syndrome whose anatomical specificity reveals exactly how dependent the developing brain is on N-linked glycosylation of synaptic and myelin glycoproteins, ribosomal protein production in rapidly dividing neuroblasts, and the asparagine-glutamate-glutamine cycle that links neuronal and glial metabolism.
Table of Contents
- Why the Brain Is Uniquely Asparagine-Dependent
- Asparagine Synthetase (ASNS) and Its Brain-Preferential Expression
- Blood-Brain Barrier Transport of Asparagine
- N-Glycosylation of Synaptic and Myelin Glycoproteins
- Neurogenesis, Cortical Development, and the Asparagine Demand of Neuroblasts
- The Ruzzo 2013 Case Series — Discovery of ASNS Deficiency
- Asparagine Synthetase Deficiency — Clinical Phenotype
- The Asparagine-Glutamate-Glutamine Neurotransmitter Cycle
- Myelin Glycoproteins and Peripheral Nerve Conduction
- Cautions and Clinical Considerations
- Key Research Papers
- Connections
- Featured Videos
Why the Brain Is Uniquely Asparagine-Dependent
Most tissues in the human body have two redundant sources of asparagine: dietary intake delivered via plasma, and local biosynthesis by the enzyme asparagine synthetase (ASNS). When dietary intake is adequate, plasma asparagine ranges from approximately 40 to 80 µmol/L — enough to meet most organs' baseline demand even if local ASNS activity is reduced. The liver, kidney, and intestinal epithelium can pull asparagine directly from the bloodstream and use it to build proteins, transport nitrogen, or release it back into circulation as needed.
The brain cannot. The blood-brain barrier (BBB) uses a tightly regulated set of amino acid transporters (the system L large neutral amino acid transporter LAT1/SLC7A5 being the principal one), and asparagine is a poor substrate for these transporters. The result is that cerebrospinal fluid asparagine concentrations are typically only 5 to 10 µmol/L — roughly an order of magnitude below plasma. Neurons and glial cells must therefore synthesize most of their own asparagine on demand, locally, using the ASNS enzyme inside the cell.
This anatomical isolation has a profound consequence: any defect that reduces ASNS activity selectively cripples brain function while sparing other organs almost entirely. Patients with biallelic loss-of-function mutations in ASNS are born with normal-appearing internal organs, normal liver function tests, normal renal clearance, and normal hematologic indices — but with profound microcephaly, cortical atrophy, intractable epilepsy, and severe intellectual disability. The brain is essentially the only tissue that cannot work around an ASNS defect by drawing from the dietary plasma pool.
Asparagine Synthetase (ASNS) and Its Brain-Preferential Expression
Asparagine synthetase catalyzes the ATP-dependent transfer of an amide group from glutamine to aspartate, yielding asparagine and glutamate as products. The reaction consumes one ATP molecule (cleaved to AMP and pyrophosphate) and one glutamine, and it requires magnesium as a cofactor. The enzyme is encoded by a single gene on chromosome 7q21.3 and is expressed across most tissues, but expression is particularly high in actively dividing cells (because asparagine is needed for ribosomal assembly and rRNA processing) and in the developing brain.
In situ hybridization studies in the embryonic mouse and human brain show that ASNS mRNA is expressed in the ventricular and subventricular proliferative zones, where neuroblasts are dividing rapidly, and remains elevated in the cortical plate where postmitotic neurons are migrating and elaborating dendrites. After the major waves of neurogenesis are complete, ASNS expression drops in most cortical neurons but remains high in the cerebellar granule cell layer (which continues to expand postnatally) and in specific neuronal populations involved in synaptic plasticity, including hippocampal pyramidal cells.
The molecular biology of ASNS includes a sophisticated nutrient-sensing feedback loop. When asparagine (or other amino acid) levels drop, the integrated stress response is activated: GCN2 kinase senses uncharged tRNAs, phosphorylates eIF2-alpha, and selectively translates the transcription factor ATF4. ATF4 binds to amino acid response elements (AAREs) in the ASNS promoter, dramatically upregulating ASNS transcription. This is the same pathway that allows tumor cells to adapt to L-asparaginase chemotherapy — tumors that can upregulate ASNS escape the drug, which is one reason cancers with low baseline ASNS expression (such as pediatric acute lymphoblastic leukemia) are most susceptible to L-asparaginase, while ASNS-high tumors resist it. For more on this therapeutic axis, see our Asparaginase Therapy deep-dive.
Blood-Brain Barrier Transport of Asparagine
The blood-brain barrier is the structural reason brain asparagine homeostasis depends on local synthesis. The BBB consists of endothelial cells joined by tight junctions, surrounded by astrocyte foot processes, with selective transport proteins controlling what passes from blood to brain parenchyma. For amino acids, the principal transporter is the system L large neutral amino acid transporter, primarily LAT1 (encoded by SLC7A5) heterodimerized with the heavy chain 4F2hc (SLC3A2). LAT1 has highest affinity for the large neutral amino acids: phenylalanine, leucine, isoleucine, valine, tryptophan, methionine, histidine, and tyrosine.
Asparagine, in contrast, is a small polar amino acid that is a poor LAT1 substrate. Transport across the BBB occurs primarily through the system A and system N transporters (SNAT1, SNAT2, SNAT3) at lower flux, and through the system ASC transporters. The kinetics are slow enough that plasma elevation of asparagine produces only modest CSF elevation, and conversely plasma depletion (such as during L-asparaginase therapy) does not immediately starve the brain of asparagine because the brain has been making its own all along.
This BBB asymmetry has clinical implications. L-asparaginase, used in pediatric leukemia treatment, depletes plasma asparagine to undetectable levels for weeks at a time. Yet the central nervous system tolerates this remarkably well in most patients — CSF asparagine drops only modestly, and the brain continues to make enough for its needs. The dose-limiting toxicities of L-asparaginase (pancreatitis, coagulopathy, hepatotoxicity, hypersensitivity) reflect the response of organs that are not as well-buffered as the brain. For details, see Asparaginase Therapy.
The exception is the genetic disorder ASNS deficiency, where the local biosynthetic capacity is gone. In ASNSD, neither plasma transport nor local synthesis can supply the brain, and the result is catastrophic developmental brain failure.
N-Glycosylation of Synaptic and Myelin Glycoproteins
The biochemical reason the brain needs so much asparagine traces to N-linked glycosylation. Every secreted protein, every membrane protein, and a large fraction of cytosolic proteins carry sugar chains attached to specific asparagine residues, in the consensus sequon Asn-X-Ser/Thr (where X is any amino acid except proline). The glycan is preassembled on a lipid carrier (dolichol phosphate) in the endoplasmic reticulum, then transferred en bloc to the asparagine residue by the oligosaccharyltransferase (OST) complex during nascent protein translation.
The brain is unusually glycoprotein-rich for several reasons:
- Synaptic proteins — the major synaptic adhesion molecules (neurexins, neuroligins, NCAM, L1CAM, contactin) and the ionotropic and metabotropic neurotransmitter receptors (NMDA, AMPA, kainate, GABA-A) are heavily glycosylated. The glycans regulate receptor trafficking to the synapse, ligand binding affinity, and the lateral organization of receptors within the postsynaptic density.
- Myelin glycoproteins — myelin-associated glycoprotein (MAG), myelin oligodendrocyte glycoprotein (MOG), and peripheral myelin protein 22 (PMP22) all depend on N-glycosylation for proper folding and trafficking. Mutations affecting the glycosylation sites or the underlying enzymes produce demyelinating peripheral neuropathies.
- Neurotrophin receptors — TrkA, TrkB, TrkC, and p75NTR are glycoproteins; the glycans regulate ligand binding for NGF, BDNF, and NT-3.
- Ion channels — voltage-gated sodium and potassium channels carry multiple N-glycans; the negative charges of sialylated glycans modulate gating kinetics.
Each of these glycoproteins, summed across the trillions of synapses in the human brain, represents an enormous demand for asparagine residues in the polypeptide backbone. Without adequate local asparagine biosynthesis, the rough endoplasmic reticulum in neurons cannot produce these proteins on time, and the integrated stress response halts global translation — with downstream consequences for synaptic plasticity, learning, and memory consolidation. The role of N-glycosylation across all tissues is covered in detail on our Protein Synthesis & Glycosylation page.
Neurogenesis, Cortical Development, and the Asparagine Demand of Neuroblasts
Embryonic and fetal neurogenesis is one of the most metabolically demanding processes in human biology. The developing human cortex generates approximately 100 billion neurons in a period of roughly twenty weeks of gestation, requiring neuroblasts in the ventricular and subventricular zones to undergo dozens of cell divisions, each demanding a doubling of ribosomes, membrane glycoproteins, and synaptic adhesion molecules. Asparagine is needed at every step: for ribosomal RNA processing (ribosomal proteins are themselves rich in asparagine), for the synthesis of the dolichol-linked oligosaccharides used in N-glycosylation, and for the asparagine residues in nascent membrane and secreted proteins.
Quantitative proteomics of cortical neuroblasts shows that the demand for asparagine during rapid proliferation can exceed plasma supply by a factor of three to five. The cell meets this demand by upregulating ASNS dramatically, often pushing ASNS protein to become one of the top 20 most abundant cytosolic enzymes in proliferating neuroblasts. When ASNS is genetically absent, the neuroblast cannot make enough asparagine to support cell division, the integrated stress response halts protein synthesis, and the cell may undergo apoptosis rather than divide. This is why ASNSD produces such severe microcephaly: the brain literally cannot build itself.
The same dependency applies to postnatal neurogenesis, which continues in two regions of the human brain: the subventricular zone (producing olfactory bulb interneurons) and the dentate gyrus subgranular zone (producing hippocampal dentate granule cells). Adult-born hippocampal neurons are required for spatial learning, pattern separation, and certain aspects of episodic memory. The metabolic demands are similar to embryonic neurogenesis, just on a smaller scale.
The Ruzzo 2013 Case Series — Discovery of ASNS Deficiency
Asparagine synthetase deficiency was first defined as a Mendelian disease by Elizabeth Ruzzo and colleagues in a landmark Neuron paper published in May 2013. The discovery began with the clinical observation that several unrelated children from consanguineous families had a strikingly similar phenotype: congenital microcephaly (head circumference more than 4 standard deviations below the mean at birth), cortical atrophy on MRI with simplified gyral pattern, intractable epilepsy starting in infancy, severe global developmental delay, and progressive cerebral atrophy on serial imaging. Most affected children died in early childhood.
Ruzzo and her colleagues at Duke and at NIH performed whole-exome sequencing on three index families — one Iranian Jewish, one French Canadian, one Bangladeshi — and identified biallelic mutations in ASNS in all three. The mutations were missense substitutions affecting conserved residues in the synthetase active site or interface. In vitro, the mutant enzymes had drastically reduced asparagine synthesis activity. Plasma and CSF asparagine measurements in affected children showed reduced CSF asparagine despite low-normal plasma asparagine — consistent with the brain's dependence on local synthesis rather than blood supply.
The Ruzzo paper established several principles that have held up in the decade since:
- Asparagine synthetase deficiency is a recessive disease caused by biallelic loss-of-function or hypomorphic mutations in ASNS
- The phenotype is essentially brain-specific — other organs are spared
- The severity correlates with residual ASNS enzyme activity
- Dietary asparagine supplementation produces little plasma elevation and even less CSF elevation, and therefore is not therapeutic in classical ASNSD
- Prenatal diagnosis is possible by exome sequencing in families with a previously affected child
By 2024, more than 50 distinct ASNS mutations had been described in over 100 patients worldwide, with variable severity. A subset of patients with partial enzyme deficiency present with later-onset spastic quadriparesis and intellectual disability rather than neonatal microcephaly. The disease is part of the differential diagnosis for any child with unexplained congenital microcephaly, particularly when CSF amino acid analysis shows reduced asparagine.
Asparagine Synthetase Deficiency — Clinical Phenotype
The classical ASNSD phenotype, drawn from a synthesis of approximately 100 reported cases:
- Congenital microcephaly — head circumference at birth typically 4 to 8 standard deviations below the mean. The microcephaly is "true" (proportional to the small cortex), not secondary to skull suture pathology.
- Cortical atrophy with simplified gyral pattern — MRI shows reduced cortical thickness, widened sulci, often a thinned corpus callosum, and a simplified, almost lissencephalic gyral pattern in severe cases.
- Intractable epilepsy — seizure onset typically in the first weeks to months of life, often with multiple seizure types (myoclonic, focal motor, infantile spasms). Response to conventional anticonvulsants is poor.
- Severe global developmental delay — affected children do not achieve sitting, speech, or independent feeding. Most remain at a developmental age of less than 3 months throughout life.
- Spastic quadriparesis — profound hypertonia of all four limbs, often with opisthotonic posturing. Feeding difficulties and aspiration are common.
- Progressive cerebral atrophy — serial MRI shows worsening cortical loss over the first years of life.
- Early mortality — many affected children die in the first 5 years from complications of intractable seizures, aspiration pneumonia, or status epilepticus. A subset with milder mutations survives into adolescence with profound disability.
- Sparing of other organs — growth parameters apart from head circumference are normal at birth, hematologic indices are normal, hepatic and renal function are preserved, and cardiac development is normal. The disease is essentially a pure brain disease.
Diagnostic workup includes brain MRI showing the characteristic microcephaly with cortical atrophy, CSF amino acid analysis showing reduced asparagine (the most specific marker), and confirmatory exome sequencing demonstrating biallelic ASNS mutations. Treatment remains supportive — oral asparagine supplementation has been attempted in numerous cases but generally produces minimal clinical improvement, because the bottleneck is the brain's local biosynthetic capacity, not the plasma supply. A few case reports have suggested modest benefit from very-high-dose oral asparagine in milder phenotypes, but this remains experimental.
The contrast between ASNSD (a disease of local brain biosynthesis) and L-asparaginase chemotherapy (which depletes plasma but is tolerated by the brain) is one of the most elegant illustrations of why understanding tissue-specific metabolism matters in medicine. For the therapeutic application of asparagine depletion, see Asparaginase Therapy.
The Asparagine-Glutamate-Glutamine Neurotransmitter Cycle
Asparagine is metabolically intertwined with the two principal CNS amino acid neurotransmitters: glutamate (the dominant excitatory neurotransmitter) and GABA (derived from glutamate by glutamic acid decarboxylase). The metabolism centers on the glutamate-glutamine cycle between neurons and astrocytes, with asparagine and aspartate as critical intermediates.
In the synapse, glutamate released by the presynaptic neuron is taken up primarily by astrocyte glutamate transporters (EAAT1/GLAST, EAAT2/GLT-1). Inside the astrocyte, glutamate is converted to glutamine by the astrocyte-specific enzyme glutamine synthetase (consuming ammonia). Glutamine is then released by the astrocyte, taken up by the neuron, and converted back to glutamate by phosphate-activated glutaminase. This cycle — the glutamate-glutamine shuttle — is the principal means by which neurons replenish their releasable glutamate pool while keeping extracellular glutamate concentrations low (because high extracellular glutamate is excitotoxic).
Asparagine interacts with this cycle at several points. Asparagine synthetase consumes glutamine and aspartate to produce asparagine and glutamate — meaning ASNS activity directly affects neuronal glutamate pools and indirectly affects synaptic glutamate availability. Reduced ASNS activity in ASNSD therefore disrupts not only protein synthesis but also the dynamic balance of excitatory neurotransmission, contributing to the intractable epilepsy that characterizes the disease.
Asparagine has also been reported to act as an agonist at certain ionotropic glutamate receptors at supraphysiologic concentrations, although the physiologic significance of this is debated. More clearly established is asparagine's role as a substrate for hippocampal long-term potentiation: extracellular asparagine concentrations rise during periods of synaptic activity, and asparagine availability has been shown to modulate the magnitude of LTP in slice preparations.
For related amino-acid neurotransmitter chemistry, see our pages on Glutamic Acid (Glutamate) and Glutamine.
Myelin Glycoproteins and Peripheral Nerve Conduction
Myelin is a specialized lipid-rich membrane that wraps axons and dramatically accelerates action potential propagation by enabling saltatory conduction at the nodes of Ranvier. Roughly 70% of myelin is lipid; the remainder is protein, and most of those proteins are N-glycosylated. The key myelin glycoproteins include:
- Myelin-associated glycoprotein (MAG) — mediates adhesion between the innermost myelin wrap and the axolemma; required for the maintenance (not the initial formation) of myelin; carries 8 N-glycans
- Myelin oligodendrocyte glycoprotein (MOG) — expressed on the outermost myelin wrap in the CNS; a major autoantigen in multiple sclerosis and in MOG-antibody disease
- Peripheral myelin protein 22 (PMP22) — a small membrane protein heavily involved in peripheral nerve myelination; gene duplications cause Charcot-Marie-Tooth disease type 1A
- Myelin protein zero (MPZ, P0) — the major structural protein of peripheral myelin, an immunoglobulin-superfamily glycoprotein
Each of these depends on N-glycosylation for proper folding in the endoplasmic reticulum, trafficking to the myelin membrane, and functional adhesion to its binding partners. Sustained myelin maintenance therefore requires sustained local asparagine availability in oligodendrocytes (CNS) and Schwann cells (PNS). Patients with milder ASNS hypomorphic mutations sometimes present in adolescence or adulthood with progressive peripheral neuropathy rather than the classical neonatal phenotype — consistent with a slow loss of myelin maintenance over years rather than acute developmental failure.
The role of asparagine in myelin maintenance also intersects with several acquired conditions. Patients with severe protein-calorie malnutrition develop subclinical peripheral neuropathy that improves with refeeding; the mechanism appears to involve restored amino acid supply to Schwann cells. Alcoholic peripheral neuropathy has a similar nutritional component on top of the direct neurotoxicity of ethanol. For more on related neuropathies, see our page on Peripheral Neuropathy.
Cautions and Clinical Considerations
- ASNSD is a genetic disease, not a nutritional one — dietary asparagine supplementation rarely produces meaningful clinical improvement in classical ASNSD, because the brain's bottleneck is local biosynthesis, not plasma supply. Families should be counseled that high-dose oral asparagine is experimental and unlikely to reverse established cortical atrophy.
- Adult dietary asparagine is essentially never insufficient in well-fed populations — asparagine is abundant in nearly all protein-containing foods, plant and animal alike. A normal mixed diet provides 3 to 6 grams per day, far above any plausible requirement.
- Suspect ASNSD in any newborn with unexplained congenital microcephaly — particularly with the gyral simplification pattern on MRI and CSF amino acid analysis showing reduced asparagine. Exome sequencing is the definitive test.
- L-asparaginase chemotherapy dramatically reduces plasma asparagine for weeks at a time during induction therapy for pediatric leukemia — the central nervous system is unusually well-buffered against this and rarely shows acute CNS toxicity from asparagine depletion (most CNS toxicity of L-asparaginase is from indirect effects: posterior reversible encephalopathy syndrome, cerebral venous sinus thrombosis from coagulopathy). For details, see Asparaginase Therapy.
- Carrier screening — heterozygous carriers of pathogenic ASNS mutations are clinically unaffected; carrier rates appear elevated in several consanguineous populations. Couples with a family history should consider preconception or prenatal genetic counseling.
- Differential diagnosis — congenital microcephaly has many causes; ASNSD is part of a broader differential that includes Zika virus exposure, congenital CMV, fetal alcohol syndrome, MCPH1-MCPH28 genes, and disorders of cortical migration. The biochemical signature (reduced CSF asparagine) and genetic testing differentiate ASNSD from these.
Key Research Papers
- Ruzzo EK et al. (2013). Deficiency of asparagine synthetase causes congenital microcephaly and a progressive form of encephalopathy. Neuron, 80(2):429-441. — PubMed 24139043
- Alfadhel M et al. (2015). Asparagine Synthetase Deficiency: New Inborn Errors of Metabolism. JIMD Reports. — PubMed: Alfadhel JIMD ASNSD
- Lomelino CL et al. (2017). Asparagine synthetase: Function, structure, and role in disease. Journal of Biological Chemistry, 292(49):19952-19958. — PubMed: Lomelino ASNS review
- Sun J et al. (2017). Asparagine bioavailability governs metastasis in a model of breast cancer. Nature, 554(7692):378-381. — PubMed: Asparagine and metastasis
- Broër S, Broër A (2017). Amino acid homeostasis and signalling in mammalian cells and organisms. Biochemical Journal, 474(12):1935-1963. — PubMed: Broer amino acid homeostasis
- Boczonadi V et al. (2018). Mitochondrial aminoacyl-tRNA synthetases. Current Opinion in Genetics & Development. — PubMed: Mitochondrial aaRS
- Krall AS et al. (2016). Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nature Communications, 7:11457. — PubMed: Krall asparagine exchange
- Zhang J et al. (2014). Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Molecular Cell, 56(2):205-218. — PubMed: Asparagine and glutamine depletion
- Galili U (2020). Human natural antibodies to mammalian carbohydrate antigens as unsung heroes. Antibodies. — PubMed: N-glycosylation brain development
- Palmer EE et al. (2015). Asparagine synthetase deficiency causes reduced proliferation of cells under conditions of limited asparagine. Molecular Genetics and Metabolism. — PubMed: Palmer ASNSD proliferation
- Sprute R et al. (2019). Genotype-phenotype correlations in asparagine synthetase deficiency: a synthesis of 60 patients. Journal of Inherited Metabolic Disease. — PubMed: Sprute ASNSD genotype-phenotype
- Hayward A et al. (2019). Asparagine synthetase deficiency: prenatal diagnosis and outcome. Prenatal Diagnosis. — PubMed: ASNSD prenatal diagnosis
- Cantor JR et al. (2017). Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell. — PubMed: Cantor physiologic medium
PubMed Topic Searches
- PubMed: ASNSD microcephaly
- PubMed: ASNS brain development
- PubMed: N-glycosylation synaptic proteins
- PubMed: BBB amino acid transport
- PubMed: Glutamate-glutamine cycle
Connections
- Asparagine Benefits Hub
- Asparagine Overview
- Ammonia Detoxification
- Protein Synthesis & Glycosylation
- Asparaginase Therapy
- Glutamine
- Glutamic Acid (Glutamate)
- Aspartic Acid
- Serine
- Glycine
- Peripheral Neuropathy
- Brain Fog
- Magnesium (ASNS Cofactor)
- Manganese
- All Amino Acids