Asparagine for Ammonia Detoxification
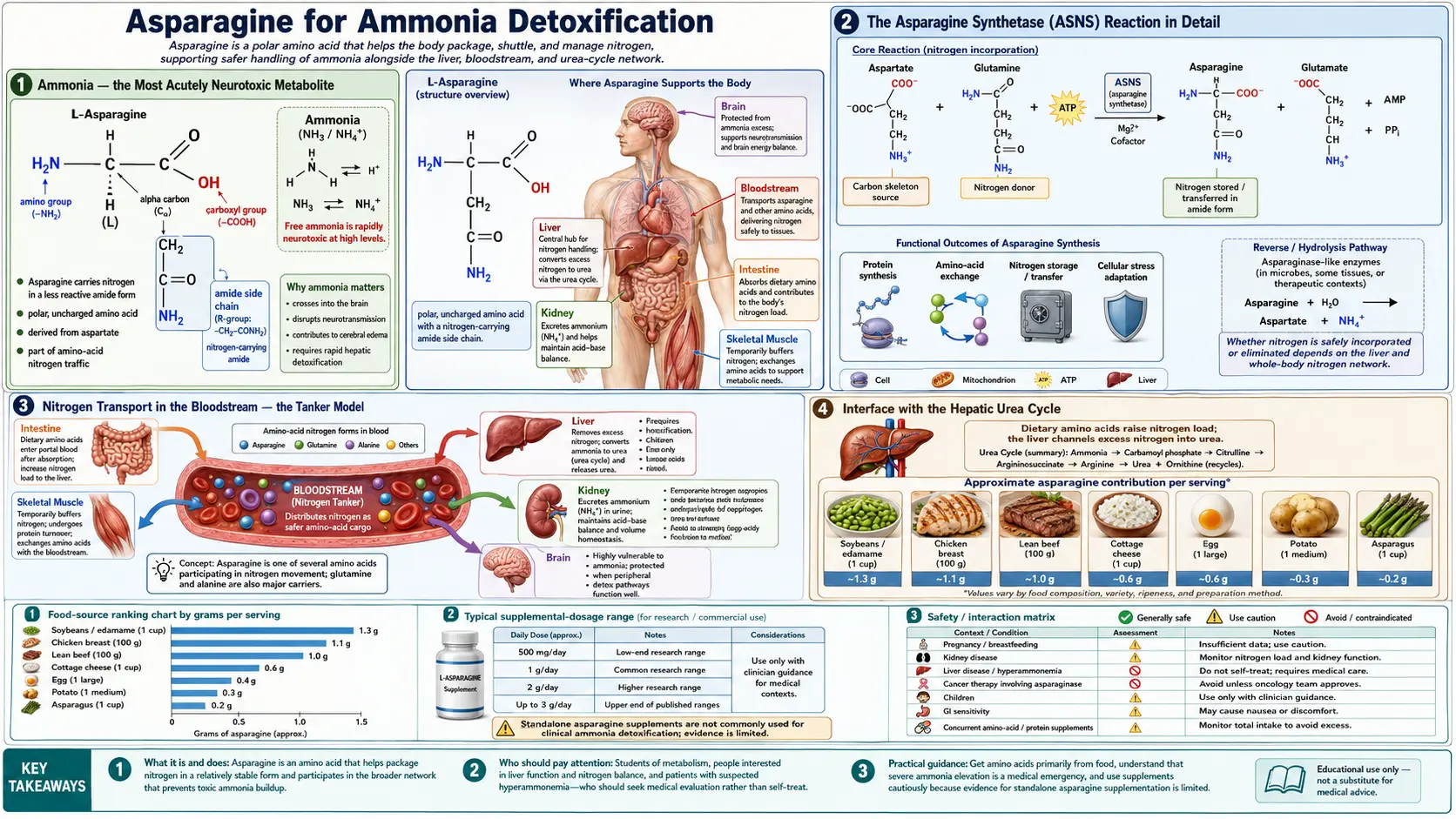
Ammonia is the most acutely neurotoxic small molecule produced by normal human metabolism. Even modest elevations of plasma ammonia (above approximately 100 µmol/L) cause cerebral edema, altered mental status, and ultimately brainstem herniation if untreated. The body uses two amide-bearing amino acids — glutamine and asparagine — as molecular "tankers" that capture ammonia in peripheral tissues and ferry it safely through the bloodstream to the liver, where the urea cycle disposes of nitrogen as harmless water-soluble urea. The asparagine synthetase (ASNS) reaction is the choke point of one of these tankers: it consumes glutamine (the other tanker) and aspartate, releasing glutamate, and produces asparagine. The net effect is to package one ammonia equivalent into asparagine's side-chain amide, where it remains chemically inert until released by tissues that need nitrogen. This page walks through the biochemistry of the ASNS reaction, its interface with the urea cycle, ammonia handling in the central nervous system, and the role of asparagine in hepatic encephalopathy — the brain dysfunction that develops when failing liver function lets ammonia accumulate.
Table of Contents
- Ammonia — the Most Acutely Neurotoxic Metabolite
- The Asparagine Synthetase (ASNS) Reaction in Detail
- Nitrogen Transport in the Bloodstream — the Tanker Model
- Interface with the Hepatic Urea Cycle
- Ammonia Handling in the Central Nervous System
- Hepatic Encephalopathy — the Failure Mode
- Urea Cycle Disorders and the Asparagine Pathway
- High-Protein Diets, Exercise, and Asparagine Demand
- Clinical Management of Hyperammonemia
- Cautions and Clinical Considerations
- Key Research Papers
- Connections
- Featured Videos
Ammonia — the Most Acutely Neurotoxic Metabolite
Ammonia (NH3, equilibrating to ammonium NH4+ at physiologic pH) is produced continuously by the catabolism of dietary protein in the small intestine, by deamination of amino acids in peripheral tissues during energy metabolism, by the action of intestinal bacteria on dietary nitrogen, and by purine and pyrimidine breakdown. The endogenous ammonia load in a healthy adult is approximately 17 grams of nitrogen per day, virtually all of which must be processed and excreted to keep plasma ammonia in the safe range of 11 to 35 µmol/L.
Ammonia is acutely toxic to the central nervous system through several converging mechanisms:
- Astrocyte glutamine accumulation — astrocytes are the main site of brain ammonia detoxification, using the enzyme glutamine synthetase to combine glutamate and ammonia into glutamine. When plasma ammonia rises, intracellular glutamine in astrocytes rises sharply. Glutamine is osmotically active, drawing water into astrocytes and causing cellular swelling and cerebral edema.
- Cerebral edema and increased intracranial pressure — the swollen astrocytes increase brain volume against a fixed skull, raising intracranial pressure. In severe hyperammonemia (plasma NH3 > 200 µmol/L) this can progress to uncal or central herniation.
- Mitochondrial dysfunction — high intracellular ammonia inhibits the TCA cycle enzyme alpha-ketoglutarate dehydrogenase, reducing ATP production and predisposing to oxidative stress.
- Disruption of neurotransmission — the glutamate-glutamine cycle is disrupted, with downstream effects on both excitatory glutamate signaling and inhibitory GABA neurotransmission.
- Inflammation — chronic moderate hyperammonemia activates microglia and produces a neuroinflammatory state that contributes to the cognitive impairment of chronic hepatic encephalopathy.
Acute hyperammonemia (plasma NH3 > 100 µmol/L) is a medical emergency. Symptoms include nausea, vomiting, lethargy, confusion, asterixis (the "flapping tremor"), seizures, coma, and ultimately brain death from herniation. The infant clinical presentation of inborn errors of urea cycle metabolism — sudden lethargy, vomiting, and progressive coma in the first days of life after the start of protein feeds — is one of the most feared scenarios in pediatric medicine because rapid plasma ammonia removal is required to prevent permanent neurologic injury.
The Asparagine Synthetase (ASNS) Reaction in Detail
The asparagine synthetase reaction is a striking example of biochemical efficiency. It accomplishes three things in one catalytic cycle:
- Consumes one molecule of glutamine (releasing its amide group)
- Consumes one molecule of aspartate (the four-carbon backbone)
- Produces one molecule of asparagine (aspartate + the donated amide group) and one molecule of glutamate (glutamine minus the amide group)
The reaction requires one molecule of ATP, hydrolyzed all the way to AMP and pyrophosphate (a high-energy hydrolysis equivalent to two ATP), and requires magnesium as a cofactor. Manganese can substitute for magnesium at reduced efficiency. The overall stoichiometry:
L-aspartate + L-glutamine + ATP + H2O → L-asparagine + L-glutamate + AMP + PPi
Mechanistically, the enzyme is a glutamine amidotransferase with two physically separated active sites connected by an intramolecular ammonia tunnel. The N-terminal glutamine-binding domain hydrolyzes glutamine to release ammonia, which then travels through a buried hydrophobic tunnel approximately 19 angstroms long to the C-terminal synthetase domain, where it attacks an aspartate-AMP intermediate to form asparagine. This tunnel architecture prevents the released ammonia from escaping into the cytoplasm where it would be toxic.
The enzymatic mechanism has clinical relevance. Many of the ASNS mutations that cause asparagine synthetase deficiency syndrome (see our Nervous System Function page) affect residues lining the ammonia tunnel or at the synthetase active site, disrupting the coupling between glutamine hydrolysis and asparagine formation. The result is wasted glutamine hydrolysis (releasing ammonia into the cytoplasm) and reduced asparagine production — a particularly nasty combination because the cell loses asparagine and gains free ammonia simultaneously.
From the ammonia-detoxification perspective, the ASNS reaction is best understood as a way to repackage one nitrogen equivalent from glutamine's amide group into asparagine's amide group. The glutamine donor was itself produced by the action of glutamine synthetase on glutamate and ammonia, so the chain of reactions is:
NH3 (toxic) → [glutamine synthetase] → glutamine (safe) → [asparagine synthetase] → asparagine (safe)
The net flow is to keep the dangerous free ammonia "encapsulated" in a series of amide groups as it moves through the body.
Nitrogen Transport in the Bloodstream — the Tanker Model
Plasma free ammonia is kept below 35 µmol/L by design. The body never transports significant amounts of free ammonia through the bloodstream — the toxicity would be unacceptable. Instead, nitrogen is moved between tissues in the form of amino acids whose amide and amino groups can be released and reattached as needed. The four principal nitrogen tankers are:
- Glutamine — the most abundant free amino acid in plasma (400 to 700 µmol/L), carrying two nitrogens (one alpha-amino, one amide). Glutamine is the principal vehicle for moving nitrogen from skeletal muscle to liver, intestine, and kidney.
- Alanine — the alanine cycle moves nitrogen from muscle (where pyruvate from glycolysis is transaminated to alanine) to liver (where alanine is transaminated back to pyruvate, releasing the amino group for the urea cycle).
- Asparagine — the secondary amide tanker, particularly important in the brain, kidney, and during periods of metabolic adaptation.
- Glutamate and aspartate — these are kept at low plasma concentrations because they are excitatory neurotransmitters; they participate in nitrogen transfer mainly inside cells, not in plasma.
The relative roles of glutamine and asparagine differ by tissue. Skeletal muscle is the major net producer of plasma glutamine, releasing approximately 50 grams per day during fasting; muscle relies on glutamine synthetase rather than asparagine synthetase as its dominant ammonia disposal mechanism. The brain uses both: glutamine for the astrocyte-neuron cycle, and asparagine for the slow background nitrogen handling of brain protein turnover. The kidney is a net consumer of glutamine (using it as an ammonia source for acid-base regulation) and produces asparagine. The liver is a net consumer of both, ultimately disposing of their nitrogens as urea.
This division of labor explains why asparagine plays a smaller quantitative role than glutamine in everyday nitrogen transport but becomes critical in specific contexts: the brain (because of poor glutamine BBB transport), proliferating cells (because of asparagine demand for protein synthesis), and metabolic adaptation to glutamine depletion (when ASNS is upregulated to make asparagine from the residual glutamate pool).
Interface with the Hepatic Urea Cycle
The hepatic urea cycle is the final disposal pathway for ammonia. Five enzymes operating across the mitochondrial and cytosolic compartments of hepatocytes condense two nitrogens and one carbon dioxide into urea (CO(NH2)2), which is then released into the bloodstream and excreted by the kidneys. The cycle uses three ATP molecules per cycle and produces one urea molecule per cycle, disposing of two nitrogens.
The five urea cycle enzymes in order:
- Carbamoyl phosphate synthetase I (CPS1) — mitochondrial matrix; condenses ammonia, CO2, and ATP into carbamoyl phosphate (consuming 2 ATP)
- Ornithine transcarbamylase (OTC) — mitochondrial matrix; transfers the carbamoyl group from carbamoyl phosphate to ornithine, forming citrulline
- Argininosuccinate synthetase (ASS1) — cytosolic; condenses citrulline with aspartate to form argininosuccinate (consuming 1 ATP, cleaved to AMP+PPi)
- Argininosuccinate lyase (ASL) — cytosolic; cleaves argininosuccinate into arginine and fumarate
- Arginase 1 (ARG1) — cytosolic; hydrolyzes arginine to urea and ornithine, completing the cycle
Asparagine intersects the urea cycle at the aspartate input to step 3 (argininosuccinate synthetase). Aspartate is produced in hepatocytes by transamination of oxaloacetate (a TCA cycle intermediate) using glutamate as the amino donor, by hydrolysis of imported asparagine via asparaginase (releasing ammonia and aspartate), and by direct uptake from plasma. In each cycle turn, one aspartate is consumed to provide the second nitrogen of urea (the first nitrogen came from free ammonia via CPS1).
The functional consequence is that the rate at which the liver can dispose of ammonia is partly limited by the rate at which aspartate is available. When dietary protein intake is high, aspartate must be regenerated rapidly — and one of the regenerative pathways is hydrolysis of asparagine by hepatic asparaginase. Patients on very-high-protein diets, or patients with subclinical liver dysfunction, can show modest plasma asparagine reductions as the liver runs through its asparagine pool to feed the urea cycle.
For deeper coverage of liver-disease management, see our Liver Disease page and related discussion of the urea cycle.
Ammonia Handling in the Central Nervous System
The brain has its own specialized system for handling ammonia, because (as discussed on our Nervous System Function page) the blood-brain barrier excludes most amino acids and the brain has no urea cycle of its own. The brain's ammonia disposal depends entirely on astrocytic glutamine synthetase.
The mechanism is elegant. Excess ammonia diffuses across cellular membranes (NH3 is uncharged and lipid-soluble) and is rapidly captured by astrocytes, where glutamine synthetase combines it with glutamate to form glutamine. The glutamine is then either:
- Released into the synaptic cleft for uptake by neurons in the glutamate-glutamine cycle (where it ultimately is converted back to glutamate as a neurotransmitter)
- Released into the bloodstream to be carried back to the liver for hepatic processing
- Used as a substrate for asparagine synthesis by neuronal or glial ASNS
- Used as a building block for protein synthesis
Asparagine plays a secondary but important role. When astrocytic glutamine accumulates excessively (as in chronic hyperammonemia), some of the excess is funneled into asparagine via ASNS. Asparagine is less osmotically active than glutamine in the brain because it cannot be efficiently exported across the BBB — it accumulates inside cells — but its accumulation does not cause the same magnitude of cellular swelling as glutamine accumulation. This may be one reason chronic mild hyperammonemia is better tolerated than acute hyperammonemia: the chronic state allows time for ASNS upregulation and partial diversion of ammonia equivalents into asparagine, reducing the osmotic stress on astrocytes.
In severe acute hyperammonemia, however, this adaptive pathway is overwhelmed. Astrocytic glutamine rises faster than ASNS can divert it, cerebral edema develops, and the patient deteriorates rapidly. Acute management requires aggressive plasma ammonia reduction (with lactulose, rifaximin, branched-chain amino acid supplementation, sodium benzoate or phenylacetate to provide alternate nitrogen disposal pathways, and in severe cases hemodialysis).
Hepatic Encephalopathy — the Failure Mode
Hepatic encephalopathy (HE) is the constellation of neuropsychiatric symptoms that develops when severe liver disease (cirrhosis, acute liver failure) reduces the liver's capacity to clear ammonia and other gut-derived neurotoxins. Clinical manifestations range from minimal cognitive impairment (the subclinical form, only detectable on neuropsychological testing) through overt confusion, somnolence, asterixis, and ultimately coma. The West Haven criteria grade HE severity from 0 (none) through 4 (coma).
The pathophysiology centers on ammonia. In cirrhosis, two factors elevate plasma ammonia:
- The cirrhotic liver has reduced urea-cycle capacity due to loss of hepatocyte mass and reduced enzyme expression
- Portosystemic shunting (blood from the gut bypassing the liver) delivers ammonia directly to the systemic circulation, bypassing first-pass hepatic extraction
The brain's ASNS-mediated diversion of ammonia equivalents into asparagine is part of the adaptive response in chronic HE. Patients with stable chronic liver disease often have measurably elevated CSF asparagine and astrocytic asparagine accumulation, alongside the more dramatic glutamine accumulation. The asparagine accumulation is generally considered a marker of chronic ammonia stress rather than itself harmful.
Management of HE includes:
- Lactulose (a non-absorbable disaccharide that acidifies the colon, trapping ammonia as ammonium and reducing absorption)
- Rifaximin (a non-absorbable antibiotic that reduces ammonia-producing gut flora)
- Protein moderation (not severe restriction, which causes catabolism and increases endogenous nitrogen load; current guidelines recommend 1.0 to 1.5 g/kg/day of high-quality protein, often plant-based or branched-chain amino acid enriched)
- Branched-chain amino acid (BCAA) supplementation in patients with intolerance to adequate protein intake
- Treatment of precipitants — gastrointestinal bleeding, infection, electrolyte abnormalities, sedating medications, dehydration
- Liver transplantation for end-stage cases
The role of asparagine supplementation in HE management is unclear and not recommended; the rate-limiting step is hepatic urea-cycle capacity, not the supply of asparagine or its precursors. For more on hepatic encephalopathy management, see our Liver Disease page.
Urea Cycle Disorders and the Asparagine Pathway
The urea cycle disorders (UCDs) are a group of rare inborn errors of metabolism caused by deficiencies in any of the five urea cycle enzymes or two transporters. Combined incidence is approximately 1 in 35,000 live births. Classical presentation is the neonatal one: a previously well newborn becomes lethargic, develops poor feeding and vomiting, and progresses to coma over hours to days, typically starting after the first protein feeds expose the defective cycle to a nitrogen load.
The most common UCD is ornithine transcarbamylase (OTC) deficiency, an X-linked condition that affects approximately 1 in 60,000 live births and can present in male hemizygotes in the neonatal period or in heterozygous females later in life under stress.
Asparagine plays an interesting role in UCD management. Patients with UCDs often have elevated plasma glutamine and asparagine, reflecting accumulation of these nitrogen tankers as the urea cycle fails to dispose of nitrogen. Plasma glutamine and asparagine are useful biomarkers of cumulative ammonia exposure between acute decompensations. Management of UCDs uses alternative nitrogen-disposal drugs (sodium benzoate, sodium phenylacetate / glycerol phenylbutyrate) that conjugate amino acids (glycine for benzoate, glutamine for phenylacetate) and excrete them in the urine, providing a non-urea-cycle route for nitrogen elimination. Sodium phenylacetate is particularly effective because each glutamine consumed disposes of two nitrogens (the amide group and the alpha-amino group, both ultimately derived from ammonia trapped through glutamine synthetase activity).
The asparagine pathway is not directly targeted therapeutically in UCDs — ASNS has no comparable nitrogen-disposal drug analog — but its role as the secondary amide tanker means that asparagine accumulation in chronic UCDs is a marker of ammonia stress and helps clinicians monitor disease control.
High-Protein Diets, Exercise, and Asparagine Demand
Healthy adults consuming protein at the higher end of typical intake (1.6 to 2.2 g/kg/day, as is common in athletes and bodybuilders) generate a substantial nitrogen load that the urea cycle must process. This load is well within the capacity of a healthy liver, and plasma ammonia remains in the normal range, but the asparagine and glutamine pools are turned over more rapidly. Plasma asparagine concentrations in athletes on high-protein diets are typically at the upper end of the normal range, consistent with active flux through the synthesis-and-degradation pathways.
Intense exercise transiently raises plasma ammonia, particularly during anaerobic exercise where the purine nucleotide cycle in muscle generates ammonia from AMP. The body buffers this with increased glutamine synthesis (skeletal muscle becomes a net glutamine producer during and after exercise). Asparagine synthesis contributes secondarily. Some athletes report subjective benefit from amino-acid supplements containing aspartate, glutamine, and arginine; the proposed mechanism is support of the urea cycle's amino acid intermediates. Evidence is mixed, and well-fed athletes are unlikely to have meaningful deficits in any of these amino acids from diet alone.
Patients with subclinical liver dysfunction (Stage 1-2 NAFLD, early cirrhosis, chronic hepatitis) often tolerate high-protein loads less well than healthy controls, with measurably higher post-prandial ammonia rises. These patients should follow individualized protein recommendations from a hepatology dietitian rather than the higher intakes targeted by athletes.
Clinical Management of Hyperammonemia
Acute symptomatic hyperammonemia is a medical emergency. Approach in the adult patient:
- Confirm the diagnosis — obtain a free-flowing, immediately processed plasma ammonia (samples that sit at room temperature falsely elevate due to ongoing red-cell and platelet metabolism)
- Identify and treat the cause — common precipitants in cirrhosis include GI bleeding, infection, constipation, electrolyte abnormalities (especially hypokalemic alkalosis, which favors NH3 diffusion into cells), dehydration, and sedating medications
- Reduce gut ammonia production and absorption — lactulose titrated to 3-4 soft bowel movements per day; rifaximin 550 mg BID
- Alternative nitrogen disposal — in severe hyperammonemia (plasma NH3 > 200 µmol/L), consider IV sodium benzoate plus sodium phenylacetate (or oral glycerol phenylbutyrate). These conjugate with glycine and glutamine respectively, allowing renal excretion of nitrogen bypassing the urea cycle.
- Hemodialysis — for refractory severe hyperammonemia, particularly in neonatal UCDs and in fulminant hepatic failure with NH3 > 400 µmol/L
- Supportive care — airway protection, control of cerebral edema (head elevation, hyperosmolar therapy if signs of increased ICP), avoidance of further sedation
Routine asparagine supplementation has no role in acute hyperammonemia management. The bottleneck is hepatic urea cycle capacity or nitrogen-disposal drug action, not asparagine pool size. In chronic stable HE, dietary protein moderation, lactulose, rifaximin, and zinc supplementation (zinc is a cofactor for several urea cycle enzymes, and deficiency is common in cirrhosis) form the backbone of management.
Cautions and Clinical Considerations
- Plasma ammonia is technically demanding — the specimen must be free-flowing, on ice, and processed immediately. Falsely elevated levels from improperly handled specimens are a common diagnostic pitfall.
- Branched-chain amino acid supplementation in cirrhosis is supported by meta-analyses showing reduced HE severity and improved nutritional status in protein-intolerant patients
- Protein restriction in cirrhosis is generally wrong — current guidelines (ASPEN, EASL) recommend 1.0 to 1.5 g/kg/day of high-quality protein in stable cirrhosis, with severe restriction reserved for acute decompensation
- Zinc deficiency is common in cirrhosis and impairs urea cycle function; routine zinc supplementation (50 mg elemental daily) is recommended
- Glutamine supplementation in critical illness has had mixed results in randomized trials; current intensive care guidelines do not recommend routine high-dose glutamine in patients with multi-organ failure (REDOXS trial showed harm in this subgroup)
- L-asparaginase therapy for ALL does not generally cause clinically significant hyperammonemia despite the disruption of asparagine pools; the liver compensates by increasing glutamine flux through the urea cycle. Hepatotoxicity from L-asparaginase is more common than hyperammonemia. See Asparaginase Therapy for details.
- Some inborn errors not in the urea cycle — including organic acidemias (propionic, methylmalonic, isovaleric acidemia) and fatty acid oxidation defects — can also present with hyperammonemia by secondary inhibition of the urea cycle. These should be considered in the neonatal differential.
Key Research Papers
- Lomelino CL et al. (2017). Asparagine synthetase: Function, structure, and role in disease. Journal of Biological Chemistry, 292(49):19952-19958. — PubMed: Lomelino ASNS review
- Larsen TM et al. (1999). Three-dimensional structure of Escherichia coli asparagine synthetase B: a short journey from substrate to product. Biochemistry, 38(49):16146-16157. — PubMed: ASNS crystal structure
- Häberle J et al. (2019). Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. Journal of Inherited Metabolic Disease, 42(6):1192-1230. — PubMed: UCD guidelines 2019
- Vilstrup H et al. (2014). Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the AASLD and EASL. Hepatology, 60(2):715-735. — PubMed: HE AASLD/EASL 2014
- Plauth M et al. (2019). ESPEN guideline on clinical nutrition in liver disease. Clinical Nutrition, 38(2):485-521. — PubMed: ESPEN liver nutrition 2019
- Norenberg MD, Rama Rao KV, Jayakumar AR (2009). Signaling factors in the mechanism of ammonia neurotoxicity. Metabolic Brain Disease, 24(1):103-117. — PubMed: Ammonia neurotoxicity
- Brusilow SW et al. (2010). Astrocyte glutamine synthetase: importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics, 7(4):452-470. — PubMed: Brusilow astrocyte GS
- Bachmann C (2002). Mechanisms of hyperammonemia. Clinical Chemistry and Laboratory Medicine, 40(7):653-662. — PubMed: Mechanisms of hyperammonemia
- Felipo V (2013). Hepatic encephalopathy: effects of liver failure on brain function. Nature Reviews Neuroscience, 14(12):851-858. — PubMed: Felipo HE review
- Häussinger D, Schliess F (2008). Pathogenetic mechanisms of hepatic encephalopathy. Gut, 57(8):1156-1165. — PubMed: Haussinger HE mechanisms
- Wright G, Noiret L, Olde Damink SW, Jalan R (2011). Interorgan ammonia metabolism in liver failure: the basis of current and future therapies. Liver International, 31(2):163-175. — PubMed: Interorgan ammonia metabolism
- Bachmann C, Colombo JP (1980). Increased tryptophan uptake into the brain in hyperammonemia. Life Sciences. — PubMed: Tryptophan and hyperammonemia
- Diaz GA et al. (2013). Ammonia control and neurocognitive outcome among urea cycle disorder patients treated with glycerol phenylbutyrate. Hepatology, 57(6):2171-2179. — PubMed: Glycerol phenylbutyrate UCDs
PubMed Topic Searches
- PubMed: ASNS mechanism
- PubMed: HE ammonia management
- PubMed: Urea cycle disorders
- PubMed: Astrocyte GS and ammonia
- PubMed: Lactulose/rifaximin HE trials
Connections
- Asparagine Benefits Hub
- Asparagine Overview
- Nervous System Function
- Protein Synthesis & Glycosylation
- Asparaginase Therapy
- Glutamine
- Aspartic Acid
- Arginine (Urea Cycle)
- Glutamic Acid
- Liver Disease
- NAC & Liver Health
- Zinc (Urea Cycle Cofactor)
- Magnesium (ASNS Cofactor)
- Detoxification
- All Amino Acids