Multiple Carboxylase Deficiency — The Inborn Errors Where Biotin Is Genuinely Lifesaving
Most pages on this site discuss biotin in the context of common adult nutrition. This page is different. Multiple carboxylase deficiency — the umbrella term for biotinidase deficiency, holocarboxylase synthetase deficiency, and the related biotin-responsive disorders — is the rare clinical territory where biotin is not a supplement but a lifesaving prescription medication. Untreated, these conditions cause seizures, deafness, optic atrophy, severe developmental delay, metabolic acidosis, and sometimes death in infancy. Treated with biotin at 5-20 mg per day for life, affected children grow up neurologically and developmentally normal. The newborn screening program that catches these patients before symptoms appear is one of the great unsung success stories of public health. This page covers the biochemistry, the clinical presentations, the screening and treatment protocols, and the related biotin-responsive basal ganglia disease.
Table of Contents
- The Biotin Cycle — The Biochemistry Made Concrete
- The Four Affected Carboxylase Enzymes
- Biotinidase Deficiency
- Holocarboxylase Synthetase Deficiency
- Biotin-Thiamine-Responsive Basal Ganglia Disease (BTBGD)
- Isolated Carboxylase Deficiencies
- Newborn Screening — A Public Health Triumph
- Treatment Protocol
- Laboratory Monitoring — And the Biotin-Test-Interference Paradox
- Patient and Family FAQ
- Cautions and Special Situations
- Key Research Papers
- Connections
- Featured Videos
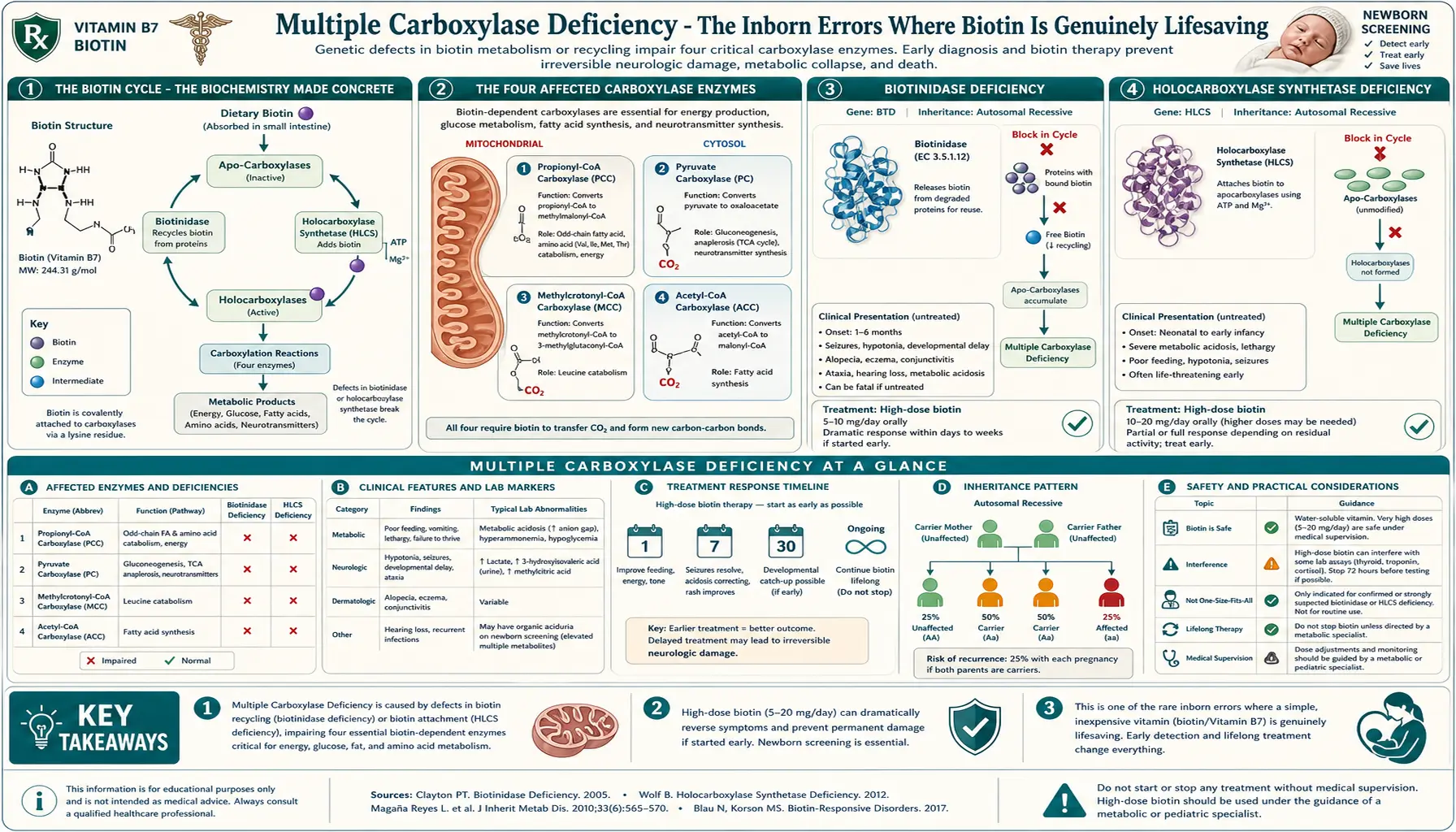
The Biotin Cycle — The Biochemistry Made Concrete
To understand the carboxylase deficiencies, you have to understand the biotin cycle. Biotin is not used and discarded; the body recycles it through every cell, every day. Two enzymes drive this cycle:
- Holocarboxylase synthetase (HCS) — the enzyme that attaches a biotin molecule to a specific lysine residue on each apocarboxylase enzyme, converting the inactive apoenzyme into the active holoenzyme. This is the activating step.
- Biotinidase — the enzyme that releases biotin back into the free pool when the carboxylase enzyme is degraded. Biotinidase cleaves the biotin-lysine bond (biocytin) and frees the biotin for reuse. This is the recycling step.
The cycle: HCS attaches biotin to apocarboxylase → carboxylase functions for the lifespan of the enzyme → enzyme is degraded by normal protein turnover → biotinidase liberates the biotin → HCS reattaches it to a new apocarboxylase. The pool of biotin available at any given moment includes dietary biotin (eaten + absorbed), intestinal-microbiome-produced biotin, and the recycled biotin from the daily turnover of cellular enzymes.
If either HCS or biotinidase is non-functional (or severely reduced in activity), the consequence is the same: failure to attach biotin to any of the carboxylase enzymes. Without biotin attached, the carboxylases do not function. The metabolic pathways they catalyze come to a halt.
The Four Affected Carboxylase Enzymes
Humans have four mitochondrial and one cytosolic biotin-dependent carboxylase enzyme. When biotin attachment fails (in either HCS or biotinidase deficiency), all four/five enzymes lose function simultaneously — hence the term multiple carboxylase deficiency:
- Pyruvate carboxylase (PC) — converts pyruvate + CO2 to oxaloacetate. Functions: (1) replenishes the citric acid cycle (anaplerosis), (2) is the first step in gluconeogenesis. Loss of function causes lactic acidosis and hypoglycemia.
- Acetyl-CoA carboxylase 1 (ACC1, cytosolic) — converts acetyl-CoA + CO2 to malonyl-CoA. Function: the first committed step of de novo fatty acid synthesis. Loss of function impairs lipid biosynthesis affecting cell membranes, myelin, and storage fat.
- Acetyl-CoA carboxylase 2 (ACC2, mitochondrial outer membrane) — same chemistry as ACC1 but localized to regulate fatty acid oxidation through malonyl-CoA inhibition of CPT-1. Loss of function dysregulates the switch between fat storage and fat burning.
- Propionyl-CoA carboxylase (PCC) — converts propionyl-CoA to methylmalonyl-CoA, which is then converted to succinyl-CoA for entry into the citric acid cycle. Substrate: propionyl-CoA derived from isoleucine, valine, methionine, threonine, odd-chain fatty acids, and cholesterol side chains. Loss of function causes propionic acid accumulation → metabolic acidosis, hyperammonemia, and propioniaciduria.
- 3-Methylcrotonyl-CoA carboxylase (MCC, also called β-methylcrotonyl-CoA carboxylase) — converts 3-methylcrotonyl-CoA to 3-methylglutaconyl-CoA in the leucine catabolism pathway. Loss of function causes accumulation of 3-methylcrotonyl-CoA and its derivatives, detectable in urine as 3-hydroxyisovaleric acid and 3-methylcrotonylglycine.
Together, these enzymes sit at the rate-limiting steps of carbohydrate (PC), fat synthesis (ACC1, ACC2), and protein catabolism (PCC, MCC). When all four/five fail simultaneously, the metabolic chaos is severe and produces the characteristic biochemical fingerprint of multiple carboxylase deficiency in newborn screening: elevated 3-hydroxyisovalerylcarnitine (C5-OH), propionylcarnitine (C3), and lactate.
Biotinidase Deficiency
Biotinidase deficiency is the more common of the two classic multiple carboxylase deficiency syndromes. It is caused by mutations in the BTD gene on chromosome 3p25 encoding the biotinidase enzyme. It is inherited as an autosomal recessive trait. Worldwide incidence is approximately 1 in 60,000 for profound deficiency (<10% residual enzyme activity) and an additional 1 in 30,000-40,000 for partial deficiency (10-30% residual activity).
Clinical presentation (untreated)
Symptoms typically develop between 3 weeks and 5 months of age, though presentation can be later in partial deficiency or in patients receiving small amounts of dietary biotin that delay overt symptoms. Without treatment, the syndrome includes:
- Cutaneous: seborrheic dermatitis, eczematoid skin rash (often around the eyes, mouth, and perineum), alopecia totalis (complete hair loss including eyebrows and eyelashes)
- Neurologic: myoclonic and tonic-clonic seizures, hypotonia, developmental delay, ataxia, cognitive regression
- Sensory: sensorineural hearing loss (often the first sign and frequently the most permanent if treatment is delayed), optic atrophy with visual loss
- Respiratory: stridor, episodes of hyperventilation, apnea (related to brainstem dysfunction)
- Metabolic: metabolic acidosis with elevated lactate, mild hyperammonemia, characteristic organic aciduria (3-hydroxyisovaleric acid, methylcrotonylglycine, 3-hydroxypropionic acid)
- Immunologic: recurrent candidal and bacterial infections (immune dysfunction)
Untreated profound biotinidase deficiency is fatal in some cases, particularly with severe metabolic decompensation triggered by intercurrent illness. The most heartbreaking aspect is that the sensorineural hearing loss and optic atrophy, once established, do not reverse with biotin treatment — making early detection imperative.
Diagnosis
- Quantitative biotinidase enzyme activity in serum (the gold standard) — profound deficiency <10%, partial 10-30%
- Urinary organic acid profile showing 3-hydroxyisovaleric aciduria with methylcrotonylglycine, 3-hydroxypropionic acid, methylcitrate
- Plasma acylcarnitine profile showing elevated C5-OH (3-hydroxyisovalerylcarnitine) and C3 (propionylcarnitine)
- Molecular confirmation by sequencing the BTD gene
- Universal newborn screening (see dedicated section below) catches presymptomatic cases
Treatment
Oral biotin, 5-20 mg per day for life. Profound deficiency: 10-20 mg daily. Partial deficiency: 5-10 mg daily. Cost is trivial — biotin is one of the cheapest pharmaceuticals in medicine. Treatment must begin as early as possible (ideally within the first weeks of life, after newborn-screening detection) to prevent the irreversible sensory damage. Once treatment begins, the cutaneous and seizure manifestations resolve within weeks. Hearing and vision, if already lost before treatment, generally do not recover, though early treatment can prevent further deterioration.
Pre-newborn-screening era cohorts (children diagnosed only after symptoms appeared) had high rates of permanent hearing loss, optic atrophy, and cognitive impairment despite eventually receiving biotin. Post-newborn-screening era cohorts (children diagnosed presymptomatically) have essentially normal outcomes. The contrast is one of the most striking demonstrations in medicine of why early detection matters.
Holocarboxylase Synthetase Deficiency
HCS deficiency is the less common but more severe of the two classic multiple carboxylase deficiencies. It is caused by mutations in the HLCS gene on chromosome 21q22.13 encoding holocarboxylase synthetase. It is also autosomal recessive. Incidence is approximately 1 in 200,000-500,000.
Clinical presentation
HCS deficiency typically presents earlier and more severely than biotinidase deficiency — classically within the first hours to days of life as overwhelming metabolic acidosis. The classic neonatal presentation:
- Severe metabolic acidosis with marked elevation of lactate, often life-threatening within hours
- Hyperammonemia
- Feeding intolerance, vomiting, lethargy progressing to coma
- Generalized erythematous scaling skin rash (often "ichthyosiform")
- Alopecia (less complete than in biotinidase deficiency)
- Seizures
- Tachypnea (respiratory compensation for the acidosis)
- Characteristic organic aciduria identical to biotinidase deficiency: 3-hydroxyisovaleric, methylcrotonylglycine, 3-hydroxypropionic, methylcitric
Some patients with HCS deficiency have a later, milder presentation in infancy or even childhood, depending on the residual activity of the mutant enzyme and the affinity of the mutant enzyme for biotin (some HCS mutations produce a Km for biotin that is markedly elevated, meaning the enzyme can still function if biotin concentration is high enough — the basis for treatment).
Diagnosis
- Plasma acylcarnitine profile (C5-OH and C3 elevated) and urinary organic acids (as in biotinidase deficiency)
- Biotinidase enzyme activity is normal (distinguishing it from biotinidase deficiency)
- Carboxylase enzyme activity assays in cultured fibroblasts show reduced activity that increases with biotin loading (functional confirmation)
- Molecular confirmation by sequencing the HLCS gene
Treatment
HCS deficiency requires higher-dose biotin than biotinidase deficiency — typically 10-80 mg per day, occasionally up to 100+ mg/day in severe cases. The rationale: most HCS mutations alter the enzyme's affinity for biotin (raise the Km) rather than abolish activity entirely. Saturating the mutant enzyme with very high substrate (biotin) concentration partially restores function. Patients with HCS mutations that destroy enzyme activity entirely do not respond fully to biotin and may have residual neurologic and metabolic issues even with treatment; patients with Km-altering mutations typically have excellent response.
Acute decompensation requires standard metabolic-emergency management: glucose to suppress catabolism, sodium bicarbonate or dialysis for severe acidosis, fluid resuscitation, and immediate high-dose biotin (oral or NG; biotin is not available IV in standard formulations).
Biotin-Thiamine-Responsive Basal Ganglia Disease (BTBGD)
BTBGD is a distinct, rarer condition that has been added to the differential of biotin-responsive disease in the past two decades. It is caused by mutations in the SLC19A3 gene encoding the thiamine transporter 2 (a transporter that, despite its name, has roles relevant to both thiamine and biotin uptake into specific tissues including the basal ganglia).
Clinical presentation
BTBGD typically presents in childhood (median age 3-10 years) with subacute encephalopathy:
- Confusion, dystonia, dysarthria, dysphagia
- Cogwheel rigidity, choreoathetosis, opisthotonus
- External ophthalmoplegia and supranuclear facial palsy
- Seizures (in a subset)
- Coma in severe untreated cases
- Episodes are often triggered by febrile illness or fasting
The characteristic MRI finding is bilateral, symmetric T2 hyperintensity in the caudate nuclei and putamina — a striking pattern that, once seen and recognized, is highly suggestive of the diagnosis. Cerebral cortical and white matter changes can also be present.
Treatment
Combination of biotin (5-10 mg/kg/day, often capped at 300 mg) plus thiamine (10-40 mg/kg/day, often 100-300 mg) for life. With early treatment, symptoms resolve dramatically over days to weeks. Delayed or untreated BTBGD progresses to permanent dystonia, intellectual disability, or death. The condition was previously diagnosed retrospectively in many cases of "Leigh-like" or unexplained encephalopathy; today, awareness of the SLC19A3 gene allows rapid genetic confirmation and early treatment.
This is a clinical scenario where empiric biotin + thiamine trial is justified in any child presenting with the characteristic MRI pattern, even before genetic testing returns — the cost is trivial, the safety profile is excellent, and the alternative (untreated progression) is catastrophic.
Isolated Carboxylase Deficiencies
Beyond the multiple carboxylase deficiencies above, several isolated single-enzyme deficiencies occur where only one carboxylase is non-functional:
- Propionic acidemia (PCC deficiency) — mutations in PCCA or PCCB genes. Presents in neonates with severe metabolic acidosis, hyperammonemia, and propionic aciduria. Does not respond to biotin (the enzyme is structurally absent, not biotin-unattached). Treatment is protein restriction, carnitine, and other metabolic measures. Distinguishing this from biotinidase/HCS deficiency requires biotinidase assay, HCS molecular testing, and confirmation by enzyme assay in cultured cells.
- Methylcrotonyl-CoA carboxylase (MCC) deficiency — mutations in MCCC1 or MCCC2. Often clinically mild, sometimes asymptomatic, identified by newborn screening with elevated C5-OH. Some cases have severe Reye-like decompensation with fasting. Does not respond to biotin in most cases.
- Pyruvate carboxylase (PC) deficiency — mutations in PC. Presents with severe lactic acidosis, hypoglycemia, neurologic deterioration in infancy. Variable severity. Does not respond to biotin in most cases.
- 3-Methylcrotonylglycinuria (transient/dietary) — some asymptomatic adults with mild C5-OH elevation on screening turn out to have no organic disease and need no treatment. This benign variant must not be confused with true MCC deficiency or with biotinidase/HCS deficiency.
The diagnostic algorithm following an abnormal newborn screen (elevated C5-OH and/or C3) is: (1) repeat the screen, (2) urinary organic acids, (3) plasma acylcarnitine profile, (4) biotinidase enzyme activity, (5) molecular testing of BTD, HLCS, SLC19A3, PCCA, PCCB, MCCC1, MCCC2, PC. Most regional metabolic centers run a coordinated workup that returns results within days to weeks.
Newborn Screening — A Public Health Triumph
Biotinidase deficiency was added to universal newborn screening panels in US states progressively from 1984 through 2013. By 2013, all 50 US states include biotinidase deficiency on their mandatory newborn-screening panel. Most developed countries have similar coverage. The screen uses a colorimetric biotinidase activity assay on the dried blood spot that every newborn provides via heel-stick in the first 24-48 hours of life.
The detection rate is excellent — profound biotinidase deficiency is reliably caught presymptomatically, allowing treatment to begin before any clinical manifestations appear. Holocarboxylase synthetase deficiency is detected indirectly through the secondary acylcarnitine abnormalities (C5-OH and C3 elevations) that the screen also captures, prompting confirmatory testing.
The public health impact is dramatic. Comparison of pre- and post-screening cohorts:
| Outcome | Pre-NBS (diagnosed after symptoms) | Post-NBS (diagnosed presymptomatically) |
|---|---|---|
| Permanent hearing loss | ~75% | <5% |
| Optic atrophy / visual impairment | ~50% | <5% |
| Developmental delay | ~60% | ~10% (often related to delay in starting treatment) |
| Seizure history | ~80% | <15% |
| Normal adult outcome on treatment | ~25% | >95% |
The screening program is one of the most cost-effective interventions in modern medicine: per dollar spent on screening, the lifetime healthcare cost avoided (preventing the lifelong support needs of a child with severe deafness, blindness, and intellectual disability) is enormous, and the human benefit is incalculable.
Treatment Protocol
Detailed treatment by syndrome:
Profound biotinidase deficiency
- Oral biotin 10-20 mg/day starting as early as possible after newborn screening confirmation (typically by day of life 7-14)
- Lifelong — never stop, never reduce. Discontinuation produces recurrence of symptoms within weeks
- Annual ophthalmology and audiology evaluation to monitor for any sensory issues
- Genetic counseling for the family — future pregnancies have 25% recurrence risk
- Sibling testing recommended even if asymptomatic (partial deficiency may be subclinical)
Partial biotinidase deficiency
- Oral biotin 5-10 mg/day; lifelong
- Some experts treat only during periods of increased risk (illness, surgery, stress); others treat universally. Practice is conservative because biotin is so safe and cheap.
Holocarboxylase synthetase deficiency
- Oral biotin 10-80 mg/day, titrated to clinical and biochemical response
- Some patients require higher doses (up to 100+ mg/day) during illness or growth spurts
- Acute decompensation: emergent metabolic stabilization (IV glucose, bicarbonate, ammonia scavengers if hyperammonemic) plus immediate high-dose biotin
- Long-term monitoring of plasma acylcarnitine profile and urinary organic acids to confirm metabolic control
- Some severe mutations are not fully biotin-responsive; protein restriction and carnitine may be needed adjuncts
BTBGD (SLC19A3)
- Biotin 5-10 mg/kg/day (up to 300 mg) PLUS thiamine 10-40 mg/kg/day (typically 100-300 mg)
- Lifelong; never stop — recurrence on discontinuation has been documented even after years of stability
- Empiric trial reasonable in any child with the characteristic bilateral basal ganglia MRI pattern, while awaiting genetic confirmation
Biotin tablets for these indications are available in 5 mg and 10 mg sizes through normal pharmaceutical channels. For higher doses, compounded preparations or accumulation of multiple tablets are used. The over-the-counter biotin supplements sold for cosmetic purposes are pharmaceutical-grade in chemistry and can be used (often more cost-effective than prescription biotin), though clinicians sometimes prefer prescription-pathway products for quality assurance.
Laboratory Monitoring — And the Biotin-Test-Interference Paradox
Patients on chronic high-dose biotin therapy for multiple carboxylase deficiency are precisely the patients most affected by the biotin lab-test interference problem. Their daily 10-80 mg biotin dose produces serum biotin concentrations far in excess of the immunoassay-interference threshold for most modern platforms.
Practical implications:
- Routine endocrine monitoring (thyroid panels, cortisol, sex hormones, vitamin D, PTH) must be done on biotin-resistant assay platforms when available, or interpreted with the interference in mind
- If acute illness develops (chest pain, abdominal pain, suspected MI, suspected sepsis), the patient and family must alert every clinician to the biotin dose. Troponin, hCG, and tumor markers can read falsely low
- Tumor surveillance in patients with the rare comorbidity of cancer plus carboxylase deficiency requires careful coordination
- Pregnancy monitoring in adult women with biotinidase deficiency (now common as the first generation of newborn-screening-detected patients reaches reproductive age) requires alternative or biotin-resistant hCG assays
For patients with multiple carboxylase deficiency, stopping the biotin to allow accurate testing is NOT an option — doing so risks acute metabolic decompensation. The lab must accommodate the patient's biotin status, not the other way around. Coordination with a metabolic specialist familiar with the patient's history is essential.
Patient and Family FAQ
Q: My newborn screen came back positive for biotinidase deficiency. What now?
The state newborn screening program will notify your pediatrician immediately. You will be referred to a metabolic specialist (typically at the nearest children's hospital). Confirmatory testing (quantitative biotinidase enzyme activity, urinary organic acids, plasma acylcarnitine profile) will be done within days to a week. If profound deficiency is confirmed, biotin treatment begins immediately at 10-20 mg/day. With prompt treatment, your child will grow up neurologically and developmentally normal.
Q: Can my child ever stop biotin?
No. Biotinidase deficiency is a permanent enzyme deficiency. Discontinuation will cause symptoms to return within weeks. Lifelong daily biotin is required. The good news: it is one of the safest, cheapest, and most effective lifelong treatments in medicine. Most patients take a single 10 mg tablet daily.
Q: Is biotin treatment dangerous?
The biotin itself is extraordinarily safe — no upper toxicity limit has been established and no direct adverse effects are seen at doses up to 300 mg/day (used in MS trials). The relevant risk is laboratory test interference at the doses your child will take. Every clinician your child sees must know about biotin and the dose; specific bloodwork may require alternative platforms or interpretation with the interference in mind. See the Lab-Test Interference page for details.
Q: My older child has hearing loss and developmental delay and was just diagnosed with biotinidase deficiency. Will biotin reverse the damage?
The metabolic and skin manifestations resolve with treatment. Unfortunately, sensorineural hearing loss and optic atrophy, once established, generally do not reverse — they can be prevented from getting worse, but the damage already done is permanent. This is why early newborn-screening detection is so important. Cognitive outcomes vary; some children recover substantially with treatment if the cognitive issues were primarily metabolic; others have persistent intellectual disability. Early intervention (PT, OT, speech therapy, audiology, ophthalmology support) is essential.
Q: Are my other children at risk?
Both parents are carriers (heterozygous) of a non-functional BTD gene. Each future pregnancy has a 25% chance of producing an affected child, 50% carrier, 25% non-carrier. Genetic counseling is recommended. Carrier testing for siblings is reasonable.
Q: Can adults develop multiple carboxylase deficiency?
Biotinidase and HCS deficiency are congenital — you are born with them. Adults who escaped diagnosis as children (rare, as newborn screening has been universal in the US since 2013) can present with adult-onset symptoms: visual or hearing problems, dermatitis, peripheral neuropathy, optic atrophy. Acquired biotin deficiency in adults (raw egg whites, anticonvulsants, TPN without biotin) produces some of the same symptoms but is biochemically distinct and reversible.
Q: What is the difference between biotinidase deficiency and HCS deficiency?
Biotinidase deficiency is more common (1 in 60,000 vs 1 in 200,000+), typically milder, presents later (weeks to months of life), and responds fully to standard biotin doses (10-20 mg/day). HCS deficiency presents earlier (often within days of birth), more severely (overwhelming metabolic acidosis), and requires higher biotin doses (10-80+ mg/day) with variable response depending on the specific mutation.
Cautions and Special Situations
- NEVER discontinue treatment. Biotinidase, HCS, and BTBGD treatments are lifelong. Discontinuation produces symptom recurrence within days to weeks, often with permanent consequences (hearing, vision, neurologic). Some patients in adulthood, feeling well, have tried to stop treatment and suffered acute decompensation.
- Lab-test interference at every encounter. The biotin dose required for these conditions (10-80 mg/day) is well above the threshold for streptavidin-biotin immunoassay interference. Every clinical encounter must include disclosure of the biotin dose and timing. See the Lab-Test Interference page for the full scope.
- Acute illness management. Intercurrent illness (viral infection, gastroenteritis, surgery) can precipitate metabolic decompensation in HCS deficiency and BTBGD. Have an emergency letter from the metabolic specialist, an "emergency protocol" outlining IV glucose and biotin top-up, and a low threshold to seek care.
- Pregnancy in affected women. Adult women with biotinidase deficiency or BTBGD can have normal pregnancies on biotin therapy. Dose may need to increase by 50-100% in the third trimester. Coordinate with both the metabolic specialist and a maternal-fetal medicine specialist. Newborn-screen the baby (the baby may be a carrier rather than affected, but the family carrier status warrants confirmation).
- Vaccinations. All standard vaccines are appropriate; biotin therapy is not an immunocompromising treatment. Some metabolic-specialist programs delay live vaccines briefly if the infant is in acute decompensation.
- Adolescent transition. Pediatric-to-adult care transition is a vulnerable period; some adolescents stop biotin out of independence-asserting impulses or because they feel well. Family + adult metabolic clinic continuity is critical.
- Distinguishing the syndromes. Multiple carboxylase deficiency due to biotinidase or HCS deficiency responds to biotin. Isolated carboxylase deficiencies (propionic acidemia, isolated MCC deficiency, isolated PC deficiency) generally do NOT respond to biotin and require different management. Accurate molecular diagnosis matters.
- BTBGD requires BOTH biotin AND thiamine. Biotin alone is insufficient; thiamine alone is insufficient; both together produce dramatic responses. This is a uniquely combined-vitamin therapy.
Key Research Papers
- Wolf B (2010). Clinical issues and frequent questions about biotinidase deficiency. Molecular Genetics and Metabolism. — PubMed
- Wolf B (2015). Why screen newborns for profound and partial biotinidase deficiency? Molecular Genetics and Metabolism. — PubMed
- Wolf B (1991). Worldwide survey of neonatal screening for biotinidase deficiency. Journal of Inherited Metabolic Disease. — PubMed
- Suormala T, Fowler B, Jakobs C et al. (1998). Late-onset holocarboxylase synthetase-deficiency. European Journal of Pediatrics. — PubMed
- Bailey LM, Ivanov RA, Wallace JC, Polyak SW (2008). Artifactual detection of biotin on histones by streptavidin. Analytical Biochemistry. — PubMed
- Tabarki B, Al-Shafi S, Al-Shahwan S et al. (2013). Biotin-responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology. — PubMed
- Ozand PT, Gascon GG, Al Essa M et al. (1998). Biotin-responsive basal ganglia disease: a novel entity. Brain. — PubMed
- Strovel ET, Cowan TM, Scott AI, Wolf B (2017). Laboratory diagnosis of biotinidase deficiency, 2017 update: a technical standard and guideline of the ACMG. Genetics in Medicine. — PubMed
- Zempleni J, Wijeratne SS, Hassan YI (2009). Biotin. BioFactors. — PubMed
- Mayr JA, Feichtinger RG, Tort F et al. (2015). Lipoic acid biosynthesis defects. Journal of Inherited Metabolic Disease. — PubMed
- Kassem H, Wafaie A, Alsuhibani S, Farid T (2014). Biotin-responsive basal ganglia disease: neuroimaging features before and after treatment. American Journal of Neuroradiology. — PubMed
- Wolf B, Spencer R, Gleason T (2002). Hearing loss is a common feature of symptomatic children with profound biotinidase deficiency. Journal of Pediatrics. — PubMed
PubMed Topic Searches
- PubMed: biotinidase deficiency
- PubMed: holocarboxylase synthetase deficiency
- PubMed: BTBGD
- PubMed: multiple carboxylase deficiency newborn screening
- PubMed: pyruvate + propionyl-CoA carboxylase
Connections
- Vitamin B7 Overview
- Benefits Hub
- Hair, Skin & Nails
- Lab-Test Interference
- High-Dose MS Trials
- Vitamin B1 (Thiamine)
- Leucine
- Isoleucine
- Valine
- Methionine
- Seizures
- Hair Loss
- Alopecia
- Seborrheic Dermatitis
- All Vitamins