Riboflavin (Vitamin B2) for MTHFR & Methylation
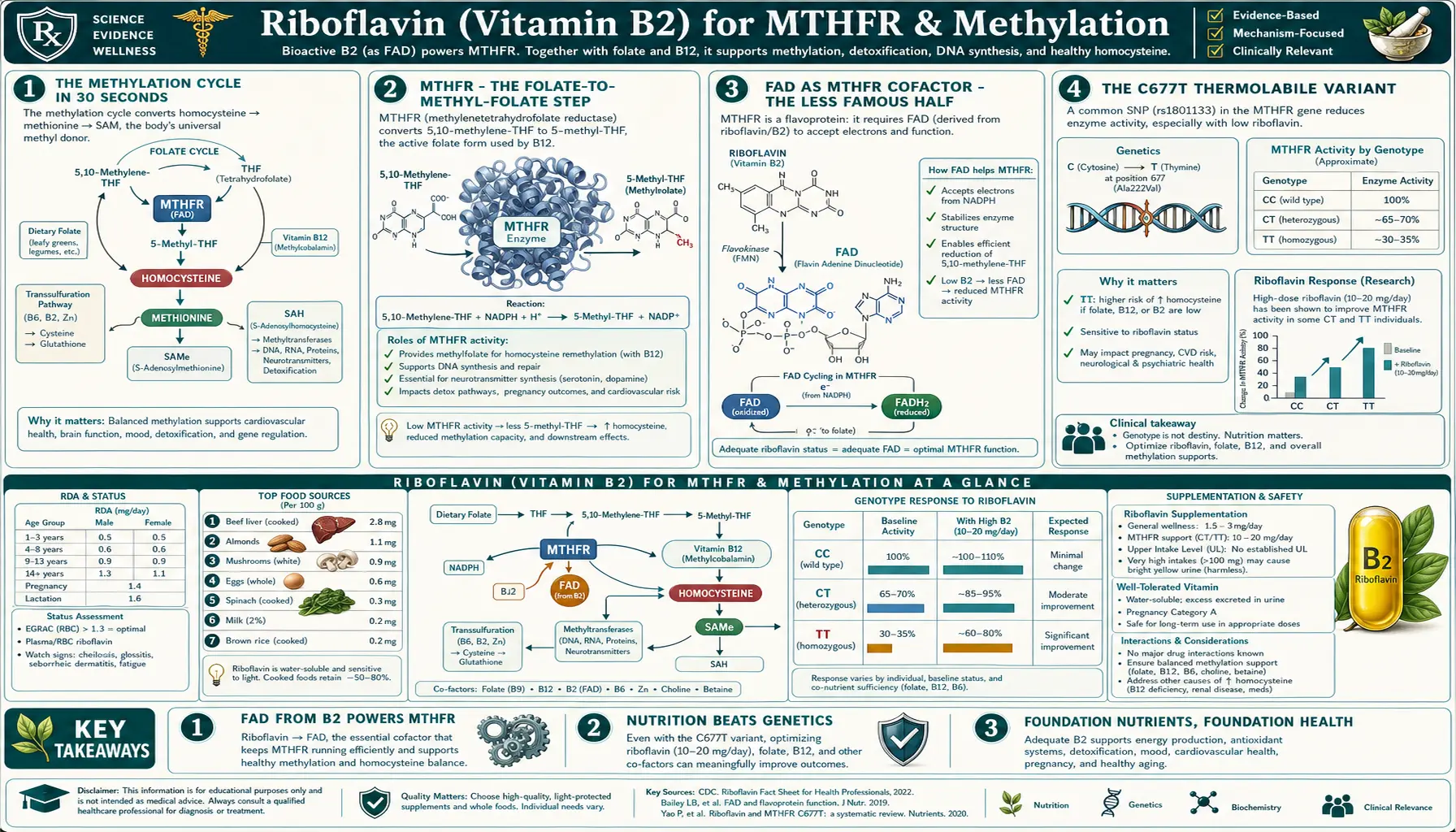
When people talk about MTHFR, they almost always talk about folate — the substrate that the enzyme acts on. The substrate matters, but the cofactor matters too, and it gets far less attention. Methylenetetrahydrofolate reductase (MTHFR) is a FAD-flavoprotein that requires riboflavin to function. In carriers of the common C677T thermolabile variant, the mutant enzyme has weaker FAD binding affinity and is unusually sensitive to riboflavin status. The McNulty, Wilson, Horigan, and Strain trials at Ulster University showed that B2 supplementation lowers homocysteine and blood pressure specifically in 677TT homozygotes — even in patients with adequate folate intake. The clinical takeaway: if you have the MTHFR C677T variant and are trying to optimize methylation, methylfolate alone is incomplete — riboflavin is the under-appreciated second cofactor that may be your rate-limiting nutrient.
Table of Contents
- The Methylation Cycle in 30 Seconds
- MTHFR — the Folate-to-Methyl-Folate Step
- FAD as MTHFR Cofactor — the Less Famous Half
- The C677T Thermolabile Variant
- Why C677T Mutants Are Riboflavin-Sensitive
- McNulty 2006 — the Homocysteine RCT
- Wilson, Horigan, and Strain — the Blood Pressure Trials
- Cardiovascular Implications of B2-MTHFR Optimization
- Bonaa NORVIT and the "Did Lowering Homocysteine Help?" Debate
- The Complete Methylation Stack
- Testing — Homocysteine, MTHFR Genotype, B2 Status
- Practical Protocol
- Cautions
- Key Research Papers
- Connections
- Featured Videos
The Methylation Cycle in 30 Seconds
Methylation is the addition of a methyl group (–CH3) to a molecule. In the body, methylation is involved in:
- DNA methylation (gene expression regulation, epigenetics)
- Neurotransmitter synthesis (serotonin, dopamine, norepinephrine, epinephrine, melatonin)
- Phospholipid synthesis (phosphatidylcholine for cell membranes and bile)
- Creatine synthesis (skeletal muscle energy)
- Detoxification (catechol-O-methyltransferase, COMT, breaks down catecholamines and estrogens)
- Homocysteine remethylation (clearing this potentially toxic intermediate)
The universal methyl donor is S-adenosylmethionine (SAMe). After donating its methyl group, SAMe becomes S-adenosylhomocysteine (SAH), which is hydrolyzed to homocysteine.
Homocysteine has two fates:
- Remethylation back to methionine, which then makes more SAMe — the cycle continues. The methyl group comes from 5-methyl-tetrahydrofolate (5-methyl-THF) via methionine synthase (B12-dependent).
- Trans-sulfuration to cysteine, which then makes taurine, sulfate, and glutathione — the "exit" from the methylation cycle into sulfur metabolism. Requires vitamin B6.
The remethylation arm of this cycle depends on a steady supply of 5-methyl-THF. That supply is generated by MTHFR.
MTHFR — the Folate-to-Methyl-Folate Step
MTHFR (methylenetetrahydrofolate reductase; gene MTHFR) reduces 5,10-methylene-tetrahydrofolate to 5-methyl-tetrahydrofolate. The methyl group on the resulting 5-methyl-THF is what eventually gets donated to homocysteine.
The reaction:
5,10-methylene-THF + NADPH → 5-methyl-THF + NADP+
(catalyzed by MTHFR, FAD-flavoprotein)
This is the committed step that diverts folate flux out of nucleotide synthesis (which needs the methylene form) and into the methylation cycle (which needs the methyl form). It is the one-way decision point in folate metabolism.
Because the reaction is essentially irreversible under cellular conditions, MTHFR activity sets the rate of methyl-folate production. Inadequate MTHFR activity means inadequate 5-methyl-THF, which means inadequate methyl donation to homocysteine, which means homocysteine accumulates in the blood — the classic biochemical signature of MTHFR dysfunction.
The standard supplement-industry response is to bypass the MTHFR step by giving preformed methylfolate (L-5-MTHF, marketed as methyl-folate, Metafolin, Quatrefolic). This works, but it doesn't address why MTHFR is underperforming in the first place. In MTHFR C677T carriers, the answer is often inadequate riboflavin.
FAD as MTHFR Cofactor — the Less Famous Half
MTHFR is a flavoprotein. Each subunit of the functional homodimer binds one FAD molecule, non-covalently but tightly. The reaction mechanism:
- NADPH docks at the enzyme's NADPH-binding site and transfers a hydride to FAD, reducing it to FADH2
- FADH2 then transfers its electrons (effectively a hydride) to 5,10-methylene-THF, reducing it to 5-methyl-THF
- The oxidized FAD is regenerated, ready for another cycle
Without FAD bound, MTHFR is catalytically inert. In riboflavin deficiency, newly synthesized MTHFR protein exists in the cell as apoenzyme (without FAD), unable to convert methylene-THF to methyl-THF, and is gradually degraded.
The clinical relevance is greatest in MTHFR C677T carriers, where the mutant enzyme has reduced FAD-binding affinity (see the next section). In wild-type (677CC) individuals, MTHFR holds onto FAD tightly enough that ordinary dietary B2 intake is enough. In 677TT homozygotes, FAD dissociates from the mutant enzyme more readily, and the cell needs an elevated FAD pool (i.e., higher riboflavin intake) to keep MTHFR adequately loaded.
This is why the MTHFR conversation that focuses only on folate is incomplete. Folate is the substrate. FAD is the cofactor. Both must be present.
The C677T Thermolabile Variant
The MTHFR C677T variant (rs1801133) is a single nucleotide polymorphism that changes a cytosine to a thymine at position 677 of the MTHFR gene. The protein-level consequence: an alanine at residue 222 becomes a valine (A222V). This valine substitution destabilizes the FAD-binding domain of the enzyme.
The variant is extremely common:
- Worldwide allele frequency: 25-30% in Europeans, 30-35% in Hispanics, 10-15% in Sub-Saharan Africans, 35-45% in some East Asian and Italian populations
- 677TT homozygotes (two copies): 10-15% of Europeans, 20-25% of Hispanics, 1-3% of Sub-Saharan Africans
- 677CT heterozygotes (one copy): 40-45% of Europeans — the most common single genotype
Frosst 1995 described the variant and named it "thermolabile" because the mutant enzyme loses activity rapidly when heated to 46°C in vitro — reflecting reduced thermal stability of the FAD-binding domain. The same destabilization causes the FAD to dissociate more easily under physiological conditions.
The clinical phenotype of 677TT homozygotes:
- Approximately 30% of wild-type MTHFR activity at body temperature
- Elevated plasma homocysteine, particularly under conditions of marginal folate or riboflavin intake
- Increased risk of certain pregnancy complications (neural tube defects, recurrent pregnancy loss, placental abruption) when folate intake is inadequate
- Slightly increased risk of certain cardiovascular and stroke outcomes in some (not all) epidemiology studies
- In riboflavin-replete populations, the phenotype is largely silent — explaining why the variant has persisted at high allele frequencies
The widespread industrial folate fortification of grain products (mandatory in the US since 1998) has substantially blunted the population-level impact of C677T on neural tube defect rates. But the variant's effect on homocysteine and methylation is not fully neutralized by folate alone — particularly in the presence of marginal riboflavin status.
Why C677T Mutants Are Riboflavin-Sensitive
The Yamada 2001 paper provided the structural-biochemical proof. Recombinant 677TT mutant MTHFR was purified and characterized:
- The mutant enzyme binds FAD with substantially lower affinity than wild type
- FAD dissociates from the mutant enzyme far more rapidly under physiological conditions
- Adding excess FAD to the in vitro reaction restores activity of the mutant enzyme toward wild-type levels
- The phenotype is rescuable in vitro by increasing FAD concentration
The translation to in vivo physiology: a 677TT homozygote whose cellular FAD pool is at the low end of the normal range will have mutant MTHFR predominantly in the apoenzyme (FAD-free) state — functionally deficient. The same person with a higher cellular FAD pool (achieved by adequate dietary riboflavin or supplementation) will have mutant MTHFR predominantly loaded with FAD — functionally restored toward normal.
This is why riboflavin supplementation in 677TT homozygotes lowers homocysteine (which depends on MTHFR-generated methyl-folate for remethylation). It is also why the effect is small or absent in 677CC wild-type individuals, who don't have a destabilized FAD-binding domain to rescue.
The fundamental insight: the C677T mutation is, biochemically, a riboflavin-dependent gain of vitamin requirement. The variant person needs more B2 than the wild-type person to make the same amount of methyl-folate. Identifying the variant via genetic testing is one way to identify who benefits most from B2 supplementation.
McNulty 2006 — the Homocysteine RCT
McNulty et al. (2006, Circulation) published the pivotal randomized trial that established B2 as a homocysteine-lowering nutrient specifically in 677TT homozygotes. The design:
- 77 adults stratified by MTHFR C677T genotype (CC, CT, TT)
- Randomized to riboflavin 1.6 mg/day or placebo for 12 weeks
- Primary outcome: plasma homocysteine
- Folate, vitamin B6, and vitamin B12 intakes were adequate and were controlled across groups
Results were striking:
- In 677TT homozygotes, plasma homocysteine fell by approximately 22% in the riboflavin group, with no change on placebo
- In 677CT heterozygotes, no significant homocysteine reduction was observed
- In 677CC wild-types, no homocysteine reduction
- The effect was specific to the 677TT genotype and was achieved at a remarkably low dose (1.6 mg/day — within the dietary range, not the megadose territory)
This trial established three things:
- Riboflavin lowers homocysteine in 677TT homozygotes
- The effect is genotype-specific — the under-the-radar mechanism most homocysteine-lowering trials miss because they don't stratify by MTHFR genotype
- Modest doses (1.6 mg/day) are sufficient — this is achievable with dietary improvement plus a B-complex; mega-doses are not required
Wilson, Horigan, and Strain — the Blood Pressure Trials
The same Ulster University group (under McNulty, Scott, Strain) extended the homocysteine work to blood pressure. The logic: 677TT homozygotes have well-documented increased blood pressure risk and increased risk of stroke. If riboflavin restores MTHFR function in these patients, blood pressure should fall.
Horigan et al. (2010, Journal of Hypertension) — 91 premature cardiovascular disease patients stratified by MTHFR genotype, given riboflavin 1.6 mg/day or placebo for 16 weeks. In the 677TT subgroup, systolic blood pressure fell by 13.2 mmHg and diastolic by 8.2 mmHg. In CC and CT subgroups, no significant change.
Wilson et al. (2013, Circulation: Cardiovascular Genetics) — 83 patients with MTHFR C677T genotype and hypertension on antihypertensive medication, given riboflavin 1.6 mg/day or placebo for 16 weeks. In 677TT homozygotes, systolic BP fell by 9.2 mmHg and diastolic by 6 mmHg over baseline. Critically, this was on top of existing antihypertensive treatment — meaning B2 supplementation has additional BP-lowering effects independent of pharmaceuticals.
Strain et al. (2017) — longer-term follow-up in 677TT homozygotes confirmed sustained blood pressure reduction over years of continued riboflavin supplementation.
The cumulative finding: in MTHFR C677T homozygotes (10-15% of the European population, more in some other ethnicities), riboflavin 1.6 mg/day reduces systolic blood pressure by approximately 9-13 mmHg. This is a clinically meaningful blood pressure reduction comparable in magnitude to single-agent antihypertensive therapy — achieved with a tiny dose of a vitamin, with no side effects, in a specific genotype subgroup that is easy to identify.
The clinical implication: any hypertensive patient should have an MTHFR genotype check; 677TT homozygotes should be supplemented with riboflavin (alongside methylfolate) as part of their hypertension management. This is one of the cleanest examples of pharmacogenomic-style personalization with a non-pharmaceutical intervention.
Cardiovascular Implications of B2-MTHFR Optimization
Elevated homocysteine is an independent risk factor for cardiovascular disease, stroke, peripheral artery disease, deep vein thrombosis, and dementia. The mechanisms include:
- Endothelial dysfunction (reduced nitric oxide bioavailability, increased oxidative stress)
- Vascular smooth muscle proliferation
- Increased thrombogenicity (platelet activation, decreased thrombomodulin)
- Disruption of methylation status of vascular regulatory genes
- Increased oxidative stress through disrupted glutathione pathways
In the MTHFR 677TT subset, homocysteine elevation is consistently observed when nutritional methylation cofactors are marginal. Riboflavin supplementation in this subset lowers homocysteine and produces a measurable blood pressure reduction. Whether this translates to fewer hard cardiovascular endpoints (heart attacks, strokes, deaths) at a population level requires longer-duration trials, but the mechanistic chain is plausible and the intermediate-endpoint evidence is robust.
The clinical practice that has emerged in cardiology integrative practice: in any patient with premature cardiovascular disease, recurrent thrombosis, unexplained hypertension, or a strong family history, check MTHFR genotype and homocysteine, and treat 677TT homozygotes with riboflavin 25-100 mg/day plus methylfolate 400-1000 mcg/day plus methylcobalamin 1000 mcg/day. The combination is inexpensive, safe, and addresses the specific upstream metabolic defect.
Bonaa NORVIT and the "Did Lowering Homocysteine Help?" Debate
Several large RCTs in the 2000s tested whether homocysteine reduction with folic acid + B12 + B6 reduces cardiovascular events in the general population:
- NORVIT (Bonaa 2006) — 3749 post-MI patients, folic acid 0.8 mg + B12 0.4 mg + B6 40 mg vs placebo. Homocysteine fell 27% in the active group. No reduction in primary cardiovascular endpoint over 40 months; in fact, a trend toward increased events in the most aggressive treatment arm.
- HOPE-2 (Lonn 2006) — 5522 high-risk patients, folic acid 2.5 mg + B6 50 mg + B12 1 mg vs placebo. Homocysteine fell, no reduction in primary endpoint.
- VISP (Toole 2004) — 3680 stroke patients. High-dose B vitamins vs low-dose. Homocysteine fell, no reduction in recurrent stroke.
The disappointing results led many clinicians to conclude that "lowering homocysteine doesn't help." That conclusion is too sweeping. Critiques:
- None of the trials stratified by MTHFR genotype. The 10-15% of patients who would benefit most (677TT homozygotes with the riboflavin-sensitive enzyme) were diluted into a population where MTHFR is fine. The genotype-stratified McNulty/Wilson/Horigan trials show specific 677TT benefit that gets washed out in unstratified pooled analyses.
- None of the trials included riboflavin in adequate doses. NORVIT, HOPE-2, and VISP gave folic acid + B6 + B12 but minimal or no riboflavin. The B2-MTHFR cofactor failure was therefore left uncorrected, and methyl-folate generation in 677TT patients remained inefficient even on high-dose folic acid.
- Synthetic folic acid (the form used in these trials) may have unintended effects — including building up unmetabolized folic acid that interferes with methylfolate transport. Modern thinking prefers methylfolate (5-MTHF) over folic acid.
- The age of intervention may matter — treating after a cardiovascular event may be too late; lifelong methylation optimization starting decades earlier may have different effects.
The takeaway for B2-MTHFR optimization: the population-wide "just lower homocysteine" strategy disappointed because it was genotype-blind and riboflavin-light. The targeted strategy — identify 677TT homozygotes, give them adequate riboflavin alongside methylfolate, B12, and B6 — addresses a specific metabolic deficit and shows measurable physiological benefit in the relevant subgroup. Whether that translates to hard endpoints in long-term genotype-stratified trials remains an open question; the mechanism and intermediate endpoints argue it should.
The Complete Methylation Stack
For optimal methylation support, particularly in MTHFR C677T carriers, the methylation cycle needs all of its cofactors and substrates:
- Methylfolate (L-5-MTHF) 400-1000 mcg/day — preformed methyl-folate that bypasses the MTHFR step
- Methylcobalamin (B12) 500-1000 mcg/day — cofactor for methionine synthase (the enzyme that uses 5-methyl-THF to remethylate homocysteine)
- Pyridoxal-5′-phosphate (active B6) 25-100 mg/day — cofactor for the trans-sulfuration arm (CBS) that diverts homocysteine to cysteine
- Riboflavin (B2) 25-100 mg/day — cofactor for MTHFR (the under-appreciated piece). Higher doses in 677TT homozygotes (50-100 mg) and during initial repletion.
- Trimethylglycine (betaine, TMG) 500-3000 mg/day — alternative methyl donor for homocysteine via the BHMT pathway; complementary to the folate-dependent route
- Zinc 15-30 mg/day — cofactor for betaine-homocysteine methyltransferase and methionine adenosyltransferase
- Choline 500-1000 mg/day — precursor to betaine (TMG); also addresses phosphatidylcholine synthesis demands of the methylation cycle
For most patients, a high-quality methylation B-complex provides folate, B12, B6, and B2 in active forms; TMG, choline, and zinc may be added separately for fuller support. The synergy is real: missing any one component creates a bottleneck.
The common over-supplementation mistake: starting on high-dose methylfolate alone (e.g., 5-15 mg/day) without B2, B12, or B6. This produces "over-methylation" symptoms (anxiety, insomnia, agitation) in some sensitive individuals because the methylation cycle is pushed without adequate downstream capacity. Layered, balanced supplementation with all cofactors prevents these effects.
Testing — Homocysteine, MTHFR Genotype, B2 Status
- Plasma total homocysteine (tHcy) — the single best functional marker of methylation cycle status. Normal < 7 µmol/L; intermediate 7-15; elevated > 15. Most labs offer this routinely.
- MTHFR C677T and A1298C genotype — one-time test; reveals whether you carry the riboflavin-sensitive variants. Available through commercial genetic services, direct-to-consumer (23andMe, AncestryDNA secondary panels), or LabCorp/Quest with physician order.
- EGRAC (erythrocyte glutathione reductase activity coefficient) — gold-standard functional B2 status marker; specialty reference labs only. Most clinicians use clinical and intake-history assessment instead.
- Plasma B12 + methylmalonic acid + folate + B6 — round out the methylation-cofactor status panel
- Yellow urine within 24-48 hours of starting riboflavin — cheapest home marker confirming B2 absorption
The minimum useful testing battery for someone investigating MTHFR-related methylation:
- MTHFR C677T + A1298C genotype (one-time)
- Plasma homocysteine at baseline and 3-6 months after intervention
- Optional: full methylation-cofactor panel (folate, B12, B6, MMA, holotranscobalamin)
Practical Protocol
For MTHFR C677T homozygotes (677TT)
- Riboflavin 25-50 mg/day (or 100 mg/day during initial repletion for 3 months, then drop to maintenance) — addresses the genotype-specific cofactor weakness
- L-5-MTHF (methylfolate) 400-1000 mcg/day — bypasses the MTHFR step
- Methylcobalamin 500-1000 mcg/day — methionine synthase cofactor
- P-5-P (pyridoxal-5′-phosphate) 25-50 mg/day — CBS cofactor for the trans-sulfuration exit
- Reassess homocysteine at 3 months; target < 7 µmol/L
- Continue indefinitely; benefits reverse on discontinuation
For MTHFR C677T heterozygotes (677CT)
- Lower-priority intervention; baseline genotype has 65% of wild-type activity
- Maintain adequate dietary B2, methylfolate, B12, B6
- If homocysteine is elevated, treat as for 677TT
For MTHFR wild-type (677CC)
- No special MTHFR-related riboflavin need; standard B-complex coverage is fine
- If homocysteine elevated despite this, look for B12 deficiency, B6 deficiency, hypothyroidism, kidney dysfunction, or other contributors
For unknown MTHFR genotype with elevated homocysteine
- Empirical methylation B-complex (with methylated forms) often resolves the issue regardless of genotype
- If response is incomplete, order MTHFR genotype to inform personalized dosing
Cautions
- Don't treat the genotype, treat the patient — an MTHFR C677T homozygote with normal homocysteine and no symptoms does not necessarily need aggressive supplementation. The variant is common, mostly silent, and not deterministic. Functional markers (homocysteine, clinical phenotype) guide treatment.
- Methylfolate sensitivity — a minority of patients (probably 5-10%) develop anxiety, insomnia, or agitation when starting methylfolate, even at low doses. Start low (200-400 mcg/day) and titrate. Adequate B2, B12, B6, and TMG prevent most over-methylation symptoms.
- Don't megadose riboflavin for MTHFR purposes — the McNulty trial showed 1.6 mg/day was sufficient. Doses of 25-100 mg/day are reasonable; 400 mg/day is the migraine dose, not the MTHFR dose.
- Be cautious of self-diagnosis from consumer genetic reports — raw genome data from 23andMe can be misinterpreted. Confirm clinically relevant findings via reputable testing.
- Pregnancy — MTHFR optimization is particularly important before and during pregnancy (neural tube defect prevention, recurrent pregnancy loss). Consult an OB/GYN or maternal-fetal medicine specialist for pregnancy-specific protocols.
- Don't stop antihypertensives without supervision — even if riboflavin lowers your BP, taper conventional antihypertensives only under physician supervision with home BP monitoring.
- Standard photosensitivity and saturable absorption cautions for riboflavin apply — store in opaque bottles, divide larger doses, take with food
Key Research Papers
- McNulty H et al. (2006). Riboflavin lowers homocysteine in individuals homozygous for the MTHFR 677C→T polymorphism. Circulation. — PubMed
- Horigan G et al. (2010). Riboflavin lowers blood pressure in cardiovascular disease patients homozygous for the 677C→T polymorphism in MTHFR. Journal of Hypertension. — PubMed
- Wilson CP et al. (2013). Blood pressure in treated hypertensive individuals with the MTHFR 677TT genotype is responsive to intervention with riboflavin. Circulation: Cardiovascular Genetics. — PubMed
- Strain JJ et al. (2017). Personalised nutrition: identifying MTHFR genotype to guide riboflavin therapy for hypertension. Proceedings of the Nutrition Society. — PubMed
- Yamada K et al. (2001). Effects of common polymorphisms on the properties of recombinant human MTHFR (FAD-binding affinity of C677T mutant). Proc Natl Acad Sci USA. — PubMed
- Frosst P et al. (1995). A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nature Genetics. — PubMed
- Hustad S et al. (2000). Riboflavin as a determinant of plasma total homocysteine: effect modification by the methylenetetrahydrofolate reductase C677T polymorphism. Clinical Chemistry. — PubMed
- Bonaa KH et al. (2006). Homocysteine lowering and cardiovascular events after acute myocardial infarction (NORVIT). New England Journal of Medicine. — PubMed
- Lonn E et al. (2006). Homocysteine lowering with folic acid and B vitamins in vascular disease (HOPE-2). New England Journal of Medicine. — PubMed
- Powers HJ (2005). Interaction among folate, riboflavin, genotype, and cancer, with reference to colorectal and cervical cancer. Journal of Nutrition. — PubMed
- Moat SJ et al. (2003). Effect of riboflavin status on the homocysteine-lowering effect of folate in relation to the MTHFR C677T genotype. Clinical Chemistry. — PubMed
- Jacques PF et al. (2002). Determinants of plasma total homocysteine concentration in the Framingham Offspring cohort. American Journal of Clinical Nutrition. — PubMed
PubMed Topic Searches
- PubMed: riboflavin MTHFR homocysteine
- PubMed: MTHFR C677T blood pressure riboflavin
- PubMed: methylation cycle cofactors
- PubMed: Ulster riboflavin MTHFR research
- PubMed: methylfolate supplementation MTHFR
Connections
- Vitamin B2 Overview
- B2 Benefits Hub
- B2 for Migraine Prevention
- B2 Mitochondrial Cofactor
- B2 Glutathione Reductase Cofactor
- Vitamin B9 (Folate)
- Folate Methylation & Homocysteine
- Folate Cardiovascular & Stroke
- Vitamin B12 (Cobalamin)
- Vitamin B6 (Pyridoxine)
- Homocysteine
- Glutathione
- Sulfur & Trans-Sulfuration
- Zinc
- Hypertension
- Riboflavin and Energy Production
- Riboflavin and Migraine Prevention