Copper Toxicity and Wilson's Disease: Symptoms, Diagnosis, and Treatment
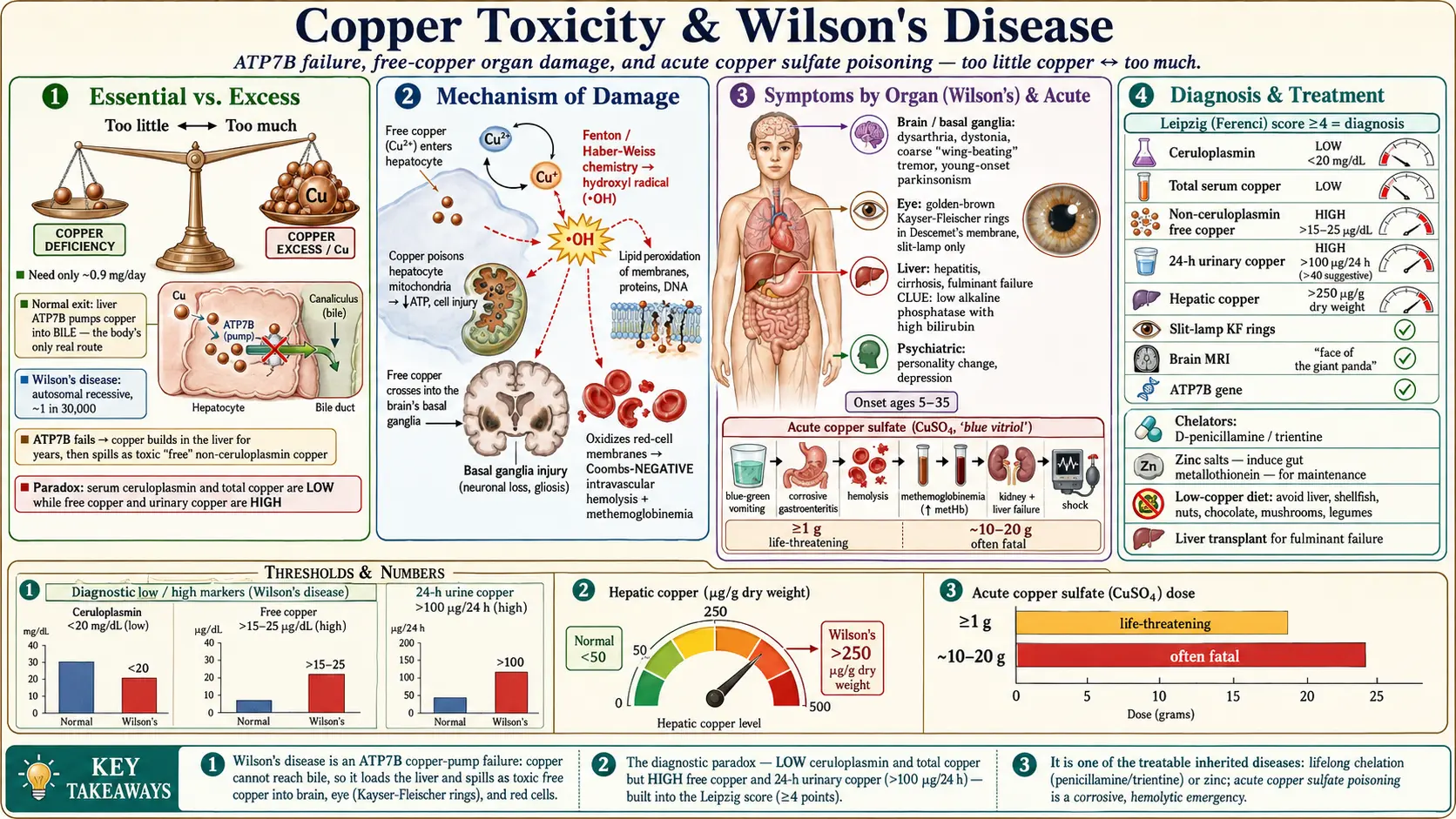
Table of Contents
- Overview
- Sources & Routes of Exposure
- Toxicokinetics
- Mechanism of Toxicity
- Symptoms & Health Effects
- Diagnosis & Laboratory Testing
- Treatment & Management
- Prevention & Risk Reduction
- Related Topics
- Key Research Papers
- Connections
- Featured Videos
1. Overview
Copper is an essential trace mineral — the body needs roughly 0.9 mg a day to run dozens of enzymes that make energy, build connective tissue, and move iron through the blood (our companion page on copper as an essential nutrient covers those beneficial roles). This page is the opposite story: what happens when the body holds too much copper. Like most minerals, copper follows a U-shaped dose curve — too little causes deficiency, the right amount is healthy, too much is a poison. Dietary copper overload is rare in healthy people, but when copper accumulates because of a broken excretion pathway, or floods in as a concentrated chemical, the consequences are severe and sometimes fatal.
The single most important cause of chronic copper toxicity is a genetic disease called Wilson's disease (also written Wilson disease, and historically hepatolenticular degeneration). An inherited fault in a copper-transport protein means the liver cannot dump excess copper into bile, so copper builds up in the liver year after year, eventually spilling into the blood as a damaging "free" form that lodges in the brain and eye. Wilson's disease is treatable — one of the few inherited metabolic diseases where lifelong therapy can produce a normal lifespan — but only if recognized. It is a great medical mimic, masquerading as ordinary hepatitis, cirrhosis, psychiatric illness, or a movement disorder, which is why it is so often diagnosed late.
The other major scenario is acute copper poisoning, almost always from swallowing a copper salt — most commonly copper sulfate, the blue compound used in agriculture and fungicides. This is a medical emergency producing corrosive gut injury, destruction of red blood cells, and liver and kidney failure within hours to days. Between these two poles sit less common chronic exposures: copper leaching from corroded plumbing into soft, acidic water; acidic foods cooked in unlined copper cookware; and infant copper-overload syndromes tied to milk stored in brass and copper vessels. This article emphasizes how copper overload presents and how it is diagnosed, because both forms are highly survivable when caught and lethal when missed — the goal here is family understanding, not self-diagnosis.
2. Sources & Routes of Exposure
Genetic accumulation: Wilson's disease
For most people who develop chronic copper toxicity, the "source" is not the environment at all — it is their own genes. In Wilson's disease, an ordinary copper-containing diet becomes dangerous over time because the body cannot get rid of the surplus. A person eating a normal Western diet takes in roughly 1–2 mg of copper a day; in health, the liver excretes the excess into bile, and it leaves in the stool. In Wilson's disease that exit route is jammed, so even a modest daily intake slowly overloads the liver. Wilson's disease is inherited in an autosomal recessive pattern, meaning a person must inherit a faulty gene copy from both parents to be affected; carriers with one faulty copy are healthy. Worldwide prevalence is on the order of 1 in 30,000, though it is higher in populations with frequent cousin marriage and in genetic isolates.
Acute chemical poisoning: copper sulfate and other salts
The most dangerous environmental copper exposure is swallowing a concentrated copper salt. Copper sulfate (CuSO₄, "blue vitriol" or "bluestone") is widely available as an agricultural fungicide, algaecide, root killer, and laboratory reagent, and its vivid blue color and easy availability make it a recurrent agent of deliberate self-poisoning, particularly in parts of South Asia where it has historically been sold cheaply and without restriction. Poisoning also occurs accidentally — in children, in industrial mishaps, from contaminated traditional remedies, and rarely from copper-contaminated dialysis fluid or intravenous solutions. The toxic threshold is low: ingestion of a gram or more of copper sulfate can be life-threatening, and amounts above roughly 10–20 grams are frequently fatal.
Chronic environmental copper
- Drinking water from corroded copper pipes. Copper plumbing can leach copper into tap water, especially where the water is soft, acidic, and standing overnight in the pipes. Water that has sat unused (first-draw morning water) and water below about pH 6.5 carries the most copper. This is a genuine but usually mild source; symptoms when they occur are gastrointestinal (nausea, cramps, diarrhea, a metallic taste).
- Unlined copper cookware. Cooking or storing acidic foods (tomato sauce, vinegar, citrus, fruit punch) in bare copper or copper-lined vessels can dissolve enough copper to cause acute gastrointestinal upset — the classic "copper colic" reported after acidic drinks served from copper containers.
- Infant copper-overload syndromes. Indian childhood cirrhosis and the closely related idiopathic copper toxicosis are severe, often fatal liver diseases of infants and toddlers historically linked to feeding milk that was boiled or stored in brass and copper vessels, probably acting together with a genetic susceptibility. These have become rare as cookware practices have changed.
Exposures that do NOT cause systemic copper toxicity
It is just as important to know what does not overload the body with copper, because copper anxiety is common and frequently misplaced. Copper intrauterine devices (IUDs) release tiny amounts of copper ions locally into the uterine cavity for contraception; the systemic copper absorbed is negligible and does not cause copper toxicity in women with normal copper metabolism. Likewise, ordinary occupational exposure to metallic copper — in plumbers, electricians, jewelers, and copper-mill workers — rarely produces systemic copper poisoning, because metallic copper and copper dust are poorly absorbed; the main occupational concern is "metal fume fever," a short-lived flu-like reaction to freshly formed copper oxide fumes, which is an inflammatory response rather than true copper overload. Copper bracelets and copper-infused fabrics deliver no meaningful systemic copper at all.
3. Toxicokinetics
How copper normally moves through the body
Wilson's disease is essentially a roadblock on the normal copper highway. Dietary copper is absorbed in the small intestine and carried to the liver bound to albumin and histidine. Inside the liver cell (hepatocyte), a copper-transporting enzyme called ATP7B does two jobs: it loads copper onto the protein ceruloplasmin, the main copper-carrier in blood, and — when copper rises — pumps the surplus into bile for excretion in the stool. Biliary excretion is the body's only significant route for getting rid of copper; the kidneys normally excrete very little.
What goes wrong in Wilson's disease
In Wilson's disease both ATP7B functions fail. Copper is not properly loaded onto ceruloplasmin, so newly made ("apo-") ceruloplasmin is unstable and is rapidly destroyed — which is why serum ceruloplasmin is typically low in Wilson's disease, and why total serum copper is also low (most blood copper is normally bound to ceruloplasmin). More importantly, copper is not pumped into bile, so it accumulates relentlessly in the liver. For years the liver acts as a sponge and the person may be entirely asymptomatic. Eventually the liver's storage capacity is overwhelmed; hepatocytes are injured and die, and they release their stored copper as a poorly-bound "free" form into the bloodstream. This non-ceruloplasmin (free) copper is the truly toxic fraction. It deposits in the basal ganglia of the brain, in the cornea (producing Kayser-Fleischer rings), and in the kidneys, and it can trigger sudden massive red-cell destruction. Because the kidney now filters this excess free copper, urinary copper excretion rises sharply — the basis of the 24-hour urinary copper test.
Acute poisoning kinetics
Acute copper-salt poisoning is faster and different. A large bolus of soluble copper overwhelms the normal binding and excretory systems within hours: it is corrosive on contact, injuring the mouth, esophagus, and stomach directly, then saturates ceruloplasmin and albumin and circulates as free copper that floods the liver and kidneys and ruptures red blood cells. Because biliary excretion cannot keep pace and the kidneys may already be failing, the toxic load is not cleared until removed by chelation.
4. Mechanism of Toxicity
Free copper and oxidative stress
The chemistry that makes copper useful is the same chemistry that makes free copper dangerous. Copper is a redox-active metal: it readily flips between its cupric (Cu²⁺) and cuprous (Cu⁺) states, donating and accepting electrons. Inside enzymes this electron-shuttling is harnessed for good. But when copper is unbound and loose in a cell, it catalyzes the Fenton and Haber-Weiss reactions, converting ordinary hydrogen peroxide into the ferociously reactive hydroxyl radical. These radicals attack whatever is nearby — cell membranes (lipid peroxidation), proteins, and DNA. The result is oxidative stress that the cell's antioxidant defenses (glutathione, metallothionein) can no longer contain.
Why the liver, brain, and red cells suffer most
Three tissues bear the brunt of copper's oxidative assault, and each failure produces a recognizable clinical picture:
- Liver. Hepatocytes are the first and largest copper store, so they are damaged first. Copper poisons their mitochondria — the cell's power plants — impairing energy production and triggering cell death. Over years this produces inflammation, fibrosis, and cirrhosis; in a sudden release it can cause acute liver failure.
- Brain. Free copper that escapes the failing liver crosses into the brain and concentrates in the basal ganglia — deep nuclei (putamen, globus pallidus, caudate) that coordinate movement. Oxidative injury and copper deposition here disrupt the fine motor control circuits, producing the tremor, rigidity, and speech and swallowing problems of neurologic Wilson's disease.
- Red blood cells. Red cells have no nucleus and limited repair capacity, so a surge of free copper oxidizes their membranes and hemoglobin and bursts them open — intravascular hemolysis. The freed hemoglobin and its breakdown can damage the kidneys, and oxidation of hemoglobin's iron produces methemoglobinemia, a form that cannot carry oxygen.
A clinically useful clue follows directly from this mechanism: because the hemolysis of Wilson's disease is caused by direct chemical (oxidative) injury rather than antibodies, it is Coombs-negative (the direct antiglobulin test is negative). A young person with unexplained Coombs-negative hemolytic anemia and liver disease should be evaluated for Wilson's disease until proven otherwise.
5. Symptoms & Health Effects
Copper toxicity does not have one face. Its symptoms depend entirely on how the copper arrived — slowly, over years (Wilson's disease), or all at once (acute poisoning). The slow form is a chronic disease of the liver, brain, and behavior; the fast form is a corrosive, hemolytic emergency.
Wilson's disease — hepatic presentation
The liver is usually affected first, and Wilson's disease can imitate almost any liver disease. Onset is typically between ages 5 and 35, though cases outside that range occur. Hepatic presentations range across a wide spectrum:
- Silent disease — only an unexplained, persistent elevation of liver enzymes (transaminases, the ALT and AST) found on a routine blood test, with no symptoms.
- Hepatitis — fatigue, poor appetite, nausea, abdominal discomfort, and jaundice (yellowing of skin and eyes), sometimes mistaken for viral or autoimmune hepatitis.
- Cirrhosis — established scarring of the liver, which may be compensated (few symptoms) or decompensated (fluid in the abdomen, easy bruising and bleeding, confusion).
- Acute (fulminant) liver failure — a catastrophic, rapidly worsening presentation, more common in young women, which carries a very high mortality without liver transplantation. Two laboratory clues strongly point to Wilson's disease as the cause: Coombs-negative hemolytic anemia occurring together with the liver failure, and a disproportionately low serum alkaline phosphatase (often with a high bilirubin), a combination fairly specific to fulminant Wilson's disease.
Wilson's disease — neurological presentation
When copper reaches the basal ganglia, a movement disorder emerges, usually in older adolescents and young adults and often after years of unrecognized liver involvement. Neurologic features include:
- Dysarthria — slurred, slow, or poorly articulated speech, frequently the earliest neurologic sign.
- Dystonia — sustained, involuntary muscle contractions that twist the limbs, neck, or face into abnormal postures; a fixed grimace ("risus sardonicus") is characteristic.
- Tremor, classically a coarse "wing-beating" tremor of the arms that appears when the arms are held outstretched, resembling the flapping of wings.
- Parkinsonism — slowness of movement, rigidity, reduced facial expression, and a shuffling gait, overlapping with Parkinson's disease but in a far younger patient.
- Drooling and difficulty swallowing, clumsiness, deteriorating handwriting, and gait or balance problems.
Wilson's disease — psychiatric presentation
Psychiatric symptoms are common and can be the very first manifestation, which is one reason young patients are sometimes treated for years for a "mental illness" before the real diagnosis is found. They include personality and behavior change, irritability and impulsivity, declining school or work performance, depression (with a meaningful suicide risk), anxiety, and, less often, frank psychosis. Because psychiatric and subtle neurologic features overlap, any young person with a new psychiatric illness plus any hint of a movement problem or liver abnormality warrants screening for Wilson's disease.
Kayser-Fleischer rings and other signs
Kayser-Fleischer (KF) rings are the visible hallmark of Wilson's disease: golden-brown to greenish rings of copper deposited in Descemet's membrane at the outer edge (limbus) of the cornea. They do not impair vision and usually cannot be seen with the naked eye — detecting them reliably requires a slit-lamp examination by an ophthalmologist. KF rings are present in the great majority of patients with neurologic Wilson's disease, but they can be absent in those with purely hepatic disease, so their absence does not rule the diagnosis out. A related but less common finding is the "sunflower cataract," a copper deposit in the lens. Other systemic effects of chronic copper overload can include kidney problems (renal tubular dysfunction, kidney stones), bone and joint disease (osteoporosis, arthritis), and episodes of hemolysis.
Acute copper sulfate poisoning
Acute ingestion of a copper salt is dramatically different — a fast-moving, multi-organ emergency. The classic sequence is:
- Corrosive gastroenteritis within minutes to hours — a metallic taste, burning of the mouth and throat, intense nausea, severe abdominal pain, and vomiting and diarrhea that are characteristically blue-green from the copper salt itself. Gastrointestinal bleeding and even perforation can occur.
- Intravascular hemolysis — the sudden bursting of red blood cells produces anemia, jaundice, and dark or red urine (hemoglobinuria).
- Methemoglobinemia — oxidized hemoglobin that cannot carry oxygen, causing bluish discoloration (cyanosis) and breathlessness that does not improve with oxygen alone.
- Liver injury — rising liver enzymes and jaundice, progressing in severe cases to acute liver failure.
- Acute kidney injury — from the combination of free copper, hemoglobin pigment, shock, and low blood pressure, sometimes requiring dialysis.
- Shock and circulatory collapse — from fluid loss, bleeding, and direct cardiovascular toxicity, the usual cause of early death.
The combination of blue-green vomit, hemolysis, and methemoglobinemia in someone who may have ingested a blue chemical is highly suggestive of copper poisoning and should prompt immediate emergency and poison-center management.
6. Diagnosis & Laboratory Testing
No single test confirms Wilson's disease; the diagnosis is built from a pattern of results. The recognized framework that combines them is the Leipzig (Ferenci) diagnostic score, agreed at the 2001 international Wilson disease meeting and endorsed in major guidelines, which assigns points for KF rings, neurologic features, ceruloplasmin, urinary and hepatic copper, the type of hemolysis, and genetic findings; a total of 4 or more points makes the diagnosis highly likely. The individual tests are described below, with the units you will see on a lab report. (Note that conventional and SI units differ; the values here are conventional U.S. units unless stated.)
Serum ceruloplasmin
Ceruloplasmin is the main copper-carrying protein in blood, and in Wilson's disease it is usually low — below about 20 mg/dL (0.2 g/L), often well below. It is a useful screening test but has important caveats. Ceruloplasmin is an acute-phase reactant, meaning it rises with inflammation, infection, pregnancy, and estrogen (oral contraceptives, hormone therapy), so a "normal" level can mask Wilson's disease during illness. Conversely, low ceruloplasmin also occurs in conditions other than Wilson's disease — severe liver failure of any cause, malnutrition, protein-losing states, and the unrelated genetic disease aceruloplasminemia. Roughly 5–15% of Wilson's patients, particularly those with hepatic disease, have ceruloplasmin in the low-normal range, so a normal value does not exclude the diagnosis.
Serum copper, total and free
Because most blood copper rides on ceruloplasmin, total serum copper is usually low in Wilson's disease — a counter-intuitive finding in a copper-overload disease. The more meaningful number is the non-ceruloplasmin ("free") copper, which is elevated and represents the toxic fraction. Free copper is estimated by subtracting the copper bound to ceruloplasmin (each mg/dL of ceruloplasmin binds about 3 micrograms of copper per dL) from the total serum copper. A free copper above roughly 15–25 µg/dL supports the diagnosis in untreated patients, and the value is also used to monitor whether treatment is working (and to detect over-treatment, which causes copper deficiency). Direct measurement of exchangeable/free copper is increasingly available and more reliable than the calculated estimate.
24-hour urinary copper
Because free copper spills into the urine, the 24-hour urinary copper excretion is one of the most useful tests. In untreated symptomatic Wilson's disease it is typically greater than 100 µg per 24 hours (1.6 µmol/24 h); a value above 40 µg/24 h is considered suggestive and warrants further work-up. Careful collection is essential (a full, copper-free container; contamination falsely raises the result). In children, in whom baseline urinary copper may be lower and less discriminating, a penicillamine challenge can be used: urine is collected over 24 hours after two oral doses of D-penicillamine, and a marked rise (historically >1600 µg/24 h) supports the diagnosis, although this cut-off is less reliable than once thought and the test is now interpreted with caution.
Slit-lamp examination for Kayser-Fleischer rings
Every patient under evaluation should have a formal slit-lamp examination by an ophthalmologist to look for KF rings, which cannot be excluded by naked-eye inspection. Their presence strongly supports the diagnosis, especially alongside neurologic disease; their absence does not exclude it, particularly in purely hepatic cases.
Brain MRI
In patients with neurologic or psychiatric features, magnetic resonance imaging (MRI) of the brain typically shows abnormal signal in the basal ganglia, thalamus, and brainstem. A well-described (though not universal) pattern in the midbrain is the "face of the giant panda" sign, created by the way copper-related signal changes outline the red nuclei and substantia nigra. MRI also helps gauge severity and track response to treatment.
Hepatic (liver) copper
Measuring copper directly in liver tissue obtained by biopsy remains a historic gold standard, especially in difficult cases. A hepatic copper concentration above 250 µg per gram of dry weight strongly supports Wilson's disease (normal is under about 50 µg/g). Because copper distribution in the liver can be patchy and other cholestatic liver diseases can also raise hepatic copper, the result is interpreted together with the rest of the picture rather than in isolation.
ATP7B genetic testing
Wilson's disease is caused by mutations in the ATP7B gene on chromosome 13, which encodes the copper-transporting protein discussed above. Over 600 disease-causing variants are known, and most patients are compound heterozygotes (carrying two different mutations), which complicates testing. Molecular genetic testing confirms the diagnosis when two pathogenic ATP7B variants are found, and is especially valuable for screening the patient's siblings — each full sibling has a 1-in-4 chance of being affected and should be tested, because pre-symptomatic treatment prevents disease. In families where the specific mutations are known, sibling screening is straightforward; otherwise, biochemical testing plus haplotype analysis is used.
Diagnosing acute copper poisoning
In a suspected acute copper-salt ingestion the diagnosis is largely clinical, supported by a markedly elevated serum copper level and the laboratory footprint of its complications: hemolysis (anemia, raised LDH and bilirubin, low haptoglobin, red-cell fragments), methemoglobinemia (measured on co-oximetry), rising liver enzymes, and acute kidney injury (rising creatinine). Management does not wait for confirmatory copper levels.
7. Treatment & Management
Treating Wilson's disease
Wilson's disease is one of the genuinely treatable inherited metabolic diseases, but treatment is lifelong — stopping it, even after years of stability, can trigger rapid, sometimes fatal relapse. The strategy is to remove the accumulated copper and then keep the body in a slightly copper-negative balance forever. Three pharmacologic tools are used, often in sequence:
- Chelating agents — D-penicillamine and trientine. These drugs bind copper and dramatically increase its excretion in the urine, pulling stored copper out of the tissues. D-penicillamine was the original oral chelator and is highly effective, but it has a substantial side-effect burden (rash, marrow suppression, kidney injury, autoimmune reactions, and a worrying early neurologic worsening in some patients with brain involvement), so it must be started cautiously and monitored closely. Trientine is an alternative chelator, generally better tolerated, and is increasingly used as first-line therapy, particularly in neurologic patients. Both require supplemental vitamin B6 considerations and careful dose titration guided by urinary copper.
- Zinc salts. Zinc works by a clever, different mechanism: it induces a copper-binding protein called metallothionein in the intestinal lining, which traps dietary and re-secreted copper inside the gut cells so that it is shed in the stool instead of absorbed. Zinc is not a fast de-copper-ing agent, but it is well tolerated and excellent for maintenance therapy and for treating pre-symptomatic patients identified by family screening. Zinc and chelators are usually given several hours apart so they do not neutralize each other.
Low-copper diet
Diet alone cannot treat Wilson's disease, but reducing copper intake supports drug therapy, especially in the first year. Patients are advised to avoid the highest-copper foods: liver and other organ meats, shellfish (especially oysters, lobster, crab), nuts and seeds, chocolate and cocoa, mushrooms, and dried legumes (beans, lentils, peas), plus high-copper tap water. Notably, these are the very same foods recommended as beneficial sources on our essential copper page — a reminder that nutrition advice is meaningless without knowing the individual's physiology.
Liver transplantation
Because the underlying defect lives in the liver, liver transplantation is effectively curative of the metabolic disease — the new liver carries normal ATP7B and restores biliary copper excretion. Transplant is reserved for acute (fulminant) liver failure from Wilson's disease and for decompensated cirrhosis that does not respond to medical therapy. It is not used for neurologic disease alone, where results are less predictable.
Monitoring
Lifelong follow-up tracks liver enzymes and function, blood counts (to catch drug-induced marrow suppression and to monitor hemolysis), and copper status — chiefly the 24-hour urinary copper and the non-ceruloplasmin free copper — to confirm the patient is neither under- nor over-treated. Over-chelation can cause iatrogenic copper deficiency, with anemia, low white cells, and a peripheral neuropathy, so the target is balance, not zero copper. Adherence is the single most important determinant of outcome.
Treating acute copper poisoning
Acute copper-salt poisoning is managed as a critical-care emergency, guided by a poison control center. The priorities are supportive and resuscitative first: securing the airway (corrosive injury and vomiting risk aspiration), aggressive intravenous fluids to counter shock and protect the kidneys, and correction of electrolyte and acid-base disturbances. Because the agent is corrosive, induced vomiting and aggressive gastric lavage are generally avoided; endoscopy may be needed to assess gut injury. Chelation (with agents such as D-penicillamine, or parenteral chelators in severe cases) is used to enhance copper elimination. The complications are treated specifically: methemoglobinemia with methylene blue when significant, severe hemolysis and anemia with transfusion and supportive care, and acute kidney injury or refractory copper levels sometimes with dialysis or extracorporeal removal. Despite optimal care, severe ingestions carry a high mortality, which is why prevention and rapid presentation matter so much.
8. Prevention & Risk Reduction
For families affected by Wilson's disease
The most powerful prevention in Wilson's disease is early detection of relatives. Because the disease is autosomal recessive, when one person is diagnosed, each of their full siblings has a 25% chance of also being affected (and a 50% chance of being a healthy carrier). First-degree relatives — especially siblings, but also children — should be screened with biochemical tests and, where the family's ATP7B mutations are known, with targeted genetic testing. Identifying and treating a relative before symptoms appear can prevent liver and brain damage entirely, turning a potentially fatal disease into a manageable one. Genetic counseling helps families understand inheritance and reproductive options.
Reducing environmental copper exposure
- Tap water. If your home has older copper plumbing and soft, acidic water, let the tap run for 30–60 seconds before drinking or cooking with the first water of the morning (standing water carries the most copper), and consider having the water tested; a certified filter or bottled water reduces intake where levels are high. This matters most for infants (whose formula is mixed with tap water) and for anyone with known or suspected Wilson's disease.
- Cookware. Do not cook or store acidic foods and drinks in unlined copper or brass vessels; choose copper cookware that is properly tin- or stainless-lined, and replace lining when it wears through.
- Chemicals. Store copper sulfate and other copper-based fungicides, algaecides, and root killers locked away from children and clearly labeled, never decanted into food or drink containers. Their attractive blue color is a particular hazard to young children. Follow label precautions when using them.
- Infant feeding. Avoid preparing or storing infant milk in brass or copper containers — the practice historically linked to fatal childhood copper-overload cirrhosis.
Sensible perspective on supplements
For the general public, dietary copper toxicity is uncommon, and most people do not need to fear ordinary copper-containing foods, copper IUDs, copper cookware that is properly lined, or copper jewelry. The recognized tolerable upper intake level for adults is around 10 mg/day from food and supplements combined — far above a typical diet. The main avoidable risks are over-supplementation (high-dose copper pills taken without need) and the rare but real danger of concentrated copper chemicals. As with every mineral on this site, the message is balance: the same copper that is essential to life becomes a poison in excess or when the body's elegant excretory machinery breaks down. Anyone with persistent unexplained liver abnormalities, a young-onset movement or psychiatric disorder, or a family history of Wilson's disease should ask a physician specifically about copper testing.
9. Related Topics
- Copper (essential mineral) — the beneficial counterpart to this page: copper's enzymes, dietary sources, and deficiency.
- Zinc — the metallothionein-inducing mineral used to treat and maintain Wilson's disease; also has its own toxicity, which interacts with copper.
- Toxic Minerals overview — the parent landing page for mineral and heavy-metal toxicity.
- Heavy Metals — shared mechanisms of oxidative metal toxicity and chelation.
- Manganism — another metal that targets the basal ganglia and mimics Parkinson's disease.
- Manganese (essential mineral) — the essential counterpart of manganism, paralleling the copper story.
- Iron — another redox-active metal whose overload damages the liver; copper and iron metabolism are closely linked through ceruloplasmin.
- Cirrhosis — a common endpoint of untreated hepatic Wilson's disease.
- Liver Disease — the broad category Wilson's disease imitates, including acute liver failure.
- Parkinson's Disease — the movement disorder neurologic Wilson's disease can mimic in young patients.
- Anemia — Coombs-negative hemolytic anemia is a key clue to Wilson's disease and acute copper poisoning.
- Lab Tests — background on the blood and urine testing used to diagnose copper toxicity.
10. Key Research Papers
- Ala A, Walker AP, Ashkan K, et al. Wilson's disease. The Lancet. 2007;369(9559):397–408. https://doi.org/10.1016/S0140-6736(07)60196-2
- Członkowska A, Litwin T, Dusek P, et al. Wilson disease. Nature Reviews Disease Primers. 2018;4(1):21. https://doi.org/10.1038/s41572-018-0018-3
- Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update (AASLD Practice Guidelines). Hepatology. 2008;47(6):2089–2111. https://doi.org/10.1002/hep.22261
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Wilson's disease. Journal of Hepatology. 2012;56(3):671–685. https://doi.org/10.1016/j.jhep.2011.11.007
- Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease (the Leipzig scoring system). Liver International. 2003;23(3):139–142. https://doi.org/10.1034/j.1600-0676.2003.00824.x
- Da Costa CM, Baldwin D, Portmann B, et al. Value of urinary copper excretion after penicillamine challenge in the diagnosis of Wilson's disease. Hepatology. 1992;15(4):609–615. https://doi.org/10.1002/hep.1840150410
- Eisenbach C, Sieg O, Stremmel W, et al. Diagnostic criteria for acute liver failure due to Wilson disease. World Journal of Gastroenterology. 2007;13(11):1711–1714. https://doi.org/10.3748/wjg.v13.i11.1711
- Appenzeller-Herzog C, Mathes T, Heeres MLS, et al. Comparative effectiveness of common therapies for Wilson disease: a systematic review and meta-analysis of controlled studies. Liver International. 2019;39(11):2136–2152. https://doi.org/10.1111/liv.14179
- Oldenquist G, Salem M. Parenteral copper sulfate poisoning causing acute renal failure. Nephrology Dialysis Transplantation. 1999;14(2):441–443. https://doi.org/10.1093/ndt/14.2.441
- Chandra A, Ansar M, Chatterjee A, et al. Fatal intravascular haemolysis due to copper sulfate poisoning: insights and literature review. BMJ Case Reports. 2024;17(12):e260160. https://doi.org/10.1136/bcr-2024-260160
- National Library of Medicine, MedlinePlus Genetics. Wilson disease. https://medlineplus.gov/genetics/condition/wilson-disease/
- Weiss KH. Wilson Disease. In: GeneReviews (NCBI Bookshelf NBK1512). University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK1512/
- National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). Wilson Disease. https://www.niddk.nih.gov/health-information/liver-disease/wilson-disease
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Copper. U.S. Department of Health and Human Services. https://www.atsdr.cdc.gov/toxprofiles/tp132.pdf
Connections
- Copper (essential mineral)
- Zinc
- Iron
- Manganese
- Toxic Minerals
- Heavy Metals
- Manganism
- Cirrhosis
- Liver Disease
- Parkinson's Disease
- Anemia
- Lab Tests
- All Minerals
- Wilson's Disease — the inherited copper-transport disorder behind most chronic copper toxicity.