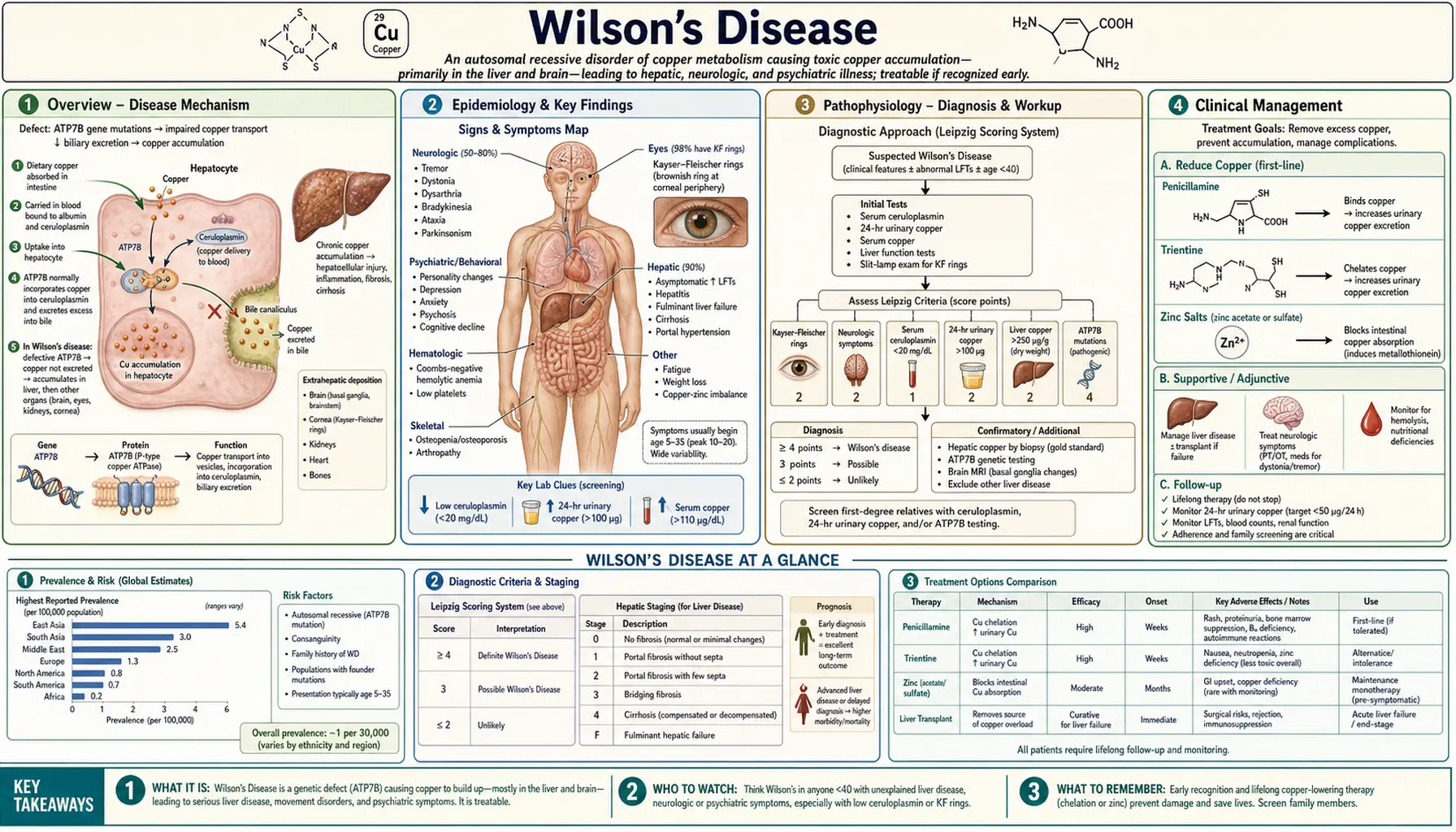
Wilson's Disease
Wilson's disease is an autosomal recessive disorder of copper metabolism caused by mutations in the ATP7B gene, which encodes a hepatic copper-transporting P-type ATPase. Defective ATP7B protein impairs biliary excretion of copper and fails to incorporate copper into ceruloplasmin, causing progressive copper accumulation in the liver, brain, cornea, kidneys, and other organs. Left untreated, Wilson's disease causes hepatic cirrhosis, neuropsychiatric deterioration, and death. With early diagnosis and lifelong copper-lowering therapy, most patients achieve normal or near-normal life expectancy.
Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention and Screening
- Recent Research and Advances
- References
1. Overview
Wilson's disease (hepatolenticular degeneration) was first described by Samuel Alexander Kinnier Wilson in 1912 as a familial progressive lenticular degeneration with cirrhosis. The disease results from pathogenic variants in ATP7B (chromosome 13q14.3), which encodes copper-transporting ATPase beta (ATP7B). This protein has two critical functions: it incorporates copper into the ferroxidase ceruloplasmin within the trans-Golgi network, and it mediates excretion of excess copper into bile. Loss of either function causes hepatic copper overload, with copper spilling into the systemic circulation and depositing in extrahepatic organs.
Wilson's disease is panethnic and affects males and females equally. Age of onset ranges from 3 to over 55 years, with most patients presenting between ages 5 and 35. The disease is uniformly fatal without treatment, but responds dramatically to copper-chelating agents (D-penicillamine, trientine) or zinc, which blocks intestinal copper absorption. Liver transplantation corrects the metabolic defect and is curative for the hepatic phenotype and most neurological manifestations.
2. Epidemiology
The worldwide prevalence of Wilson's disease is estimated at 1 in 30,000, with a carrier frequency of approximately 1 in 90 in the general population. Over 700 distinct pathogenic ATP7B mutations have been catalogued; no single mutation accounts for the majority of cases in most populations. The p.His1069Gln mutation (c.3207C>A) is the most common variant in European and North American patients, present in approximately 35–45% of alleles in those populations. The p.Arg778Leu mutation predominates in East Asian populations.
Wilson's disease affects all ethnic groups without significant sex predilection. The disease is among the most common inherited metabolic liver diseases and is an important, treatable cause of liver disease in children and young adults. In Western series, approximately 40–50% of patients present with primarily hepatic disease, 40–50% with primarily neuropsychiatric disease, and 10% with both.
3. Pathophysiology
Copper Metabolism and ATP7B Function
Dietary copper (1.0–1.5 mg/day absorbed) is taken up by enterocytes via the copper transporter CTR1, delivered to the portal circulation, and extracted by hepatocytes. Within hepatocytes, copper is distributed by copper chaperones: ATOX1 delivers copper to ATP7B in the trans-Golgi network (TGN). Under normal copper conditions, ATP7B resides in the TGN, where it loads copper onto apoceruloplasmin to form holocene ceruloplasmin. When hepatocellular copper is high, ATP7B traffics to the bile canalicular membrane to pump excess copper into bile for fecal excretion — the primary route of copper homeostasis.
In Wilson's disease, mutant ATP7B fails at one or both functions. The result is:
- Reduced ceruloplasmin synthesis: Apoceruloplasmin without copper is rapidly degraded; serum ceruloplasmin falls below 20 mg/dL in approximately 80% of patients
- Impaired biliary copper excretion: Hepatic copper accumulates, initially in periportal hepatocytes, causing steatosis and inflammation
- Copper redistribution: As hepatic storage capacity is exceeded, non-ceruloplasmin-bound copper (free copper) spills into the blood, depositing in the brain (basal ganglia, thalamus, brainstem), cornea (Descemet's membrane), renal tubules, red blood cells (causing hemolysis), and other organs
Mechanisms of Tissue Injury
Copper toxicity operates through multiple mechanisms: generation of reactive oxygen species (ROS) via Fenton-type reactions (Cu⁺ + H₂O₂ → Cu²⁺ + OH⁻ + OH·); mitochondrial dysfunction (copper accumulates in mitochondria, impairing oxidative phosphorylation); and direct protein oxidation and lipid peroxidation. In the liver, this produces a spectrum from steatohepatitis to acute liver failure with Coombs-negative hemolytic anemia (due to massive copper release from necrotic hepatocytes lysing red blood cells) to micronodular cirrhosis. In the brain, copper deposition in the lenticular nuclei (putamen and globus pallidus) causes the characteristic basal ganglia syndrome.
4. Clinical Presentation
Hepatic Wilson's Disease
Liver disease is the presenting manifestation in approximately 40–50% of patients and typically appears in the first two decades of life, often before neurological symptoms. Presentations include:
- Asymptomatic hepatomegaly or elevated aminotransferases: Discovered incidentally; the most common initial finding in children
- Chronic hepatitis: Mimics autoimmune hepatitis with elevated AST/ALT, fatigue, and jaundice; may be misdiagnosed as AIH
- Cirrhosis: Present in up to 40% at diagnosis; may be compensated (splenomegaly, thrombocytopenia) or decompensated (ascites, varices, encephalopathy)
- Acute liver failure (ALF) with Coombs-negative hemolytic anemia: The most dramatic and life-threatening hepatic presentation; characterized by rapid onset jaundice, coagulopathy, renal failure, and intravascular hemolysis (due to massive copper release from the necrotic liver); paradoxically, serum ceruloplasmin and aminotransferases may be only modestly elevated because functional hepatic mass is depleted; alkaline phosphatase is characteristically low or normal in Wilsonian ALF — an important diagnostic clue. This presentation requires emergent liver transplantation.
Neuropsychiatric Wilson's Disease
Neurological disease typically presents in the second to third decade of life, almost always after some degree of hepatic copper deposition. The neurological phenotype spans two broad patterns:
- Movement disorder (dyskinetic) phenotype: Tremor (intention tremor, "wing-beating" tremor with arms abducted), dysarthria (scanning or hypophonic speech), dysphagia, incoordination, and rigidity. Basal ganglia involvement causes bradykinesia similar to Parkinsonism. Seizures occur in approximately 10%.
- Psychiatric phenotype: Behavioral changes (irritability, impulsivity, disinhibition), personality change, depression, anxiety, and frank psychosis. Psychiatric symptoms often precede the diagnosis by years. Personality changes in a young person with any liver abnormality should trigger Wilson's evaluation.
A Wilson's disease score (Leipzig score) ≥4 points supports the diagnosis; scoring incorporates Kayser-Fleischer rings, neurological symptoms, ceruloplasmin, Coombs-negative hemolysis, liver biopsy copper, urine copper, and ATP7B mutation analysis.
Kayser-Fleischer Rings
Kayser-Fleischer (KF) rings are golden-brown to greenish deposits of copper in the peripheral cornea (Descemet's membrane), best seen at the superior and inferior poles before becoming circumferential. They are pathognomonic of Wilson's disease when present in the appropriate clinical context, but are absent in approximately 50% of patients with purely hepatic Wilson's disease and in most presymptomatic individuals. KF rings are present in virtually all patients with neurological Wilson's disease. Detection requires slit-lamp examination by an experienced ophthalmologist — they are rarely visible to the naked eye. KF rings can also be seen in other causes of prolonged cholestasis (primary biliary cholangitis, primary sclerosing cholangitis) but are rare outside Wilson's disease. Sunflower cataracts (copper deposits in the lens) are less common but also pathognomonic.
Other Manifestations
- Renal: Fanconi syndrome (proximal tubular dysfunction causing aminoaciduria, glycosuria, phosphaturia, uricosuria, renal tubular acidosis); nephrolithiasis; hematuria
- Skeletal: Osteoporosis, osteomalacia, spontaneous fractures, premature osteoarthritis
- Cardiac: Cardiomyopathy and arrhythmias (rare; more common in advanced disease)
- Endocrine: Hypoparathyroidism, delayed puberty, menstrual irregularities, recurrent spontaneous abortion
- Hematologic: Coombs-negative hemolytic anemia (the hallmark of fulminant presentation)
5. Diagnosis
Biochemical Tests
- Serum ceruloplasmin: The most commonly used screening test. Values below 20 mg/dL are found in approximately 80% of patients with Wilson's disease. However, ceruloplasmin is an acute-phase reactant and may be normal or near-normal in acute liver inflammation or during pregnancy. Conversely, low ceruloplasmin is not specific for Wilson's disease — it also occurs in other protein-losing states (Menkes disease, nephrotic syndrome, severe malnutrition, aceruloplasminemia). A normal ceruloplasmin does not exclude Wilson's disease, particularly in hepatic presentations.
- 24-hour urine copper: A value >100 mcg/day (1.6 µmol/day) is considered the diagnostic threshold in symptomatic patients. Values >40 mcg/day are suspicious. In asymptomatic first-degree relatives, the D-penicillamine challenge test (500 mg D-penicillamine orally at 0 and 12 hours, with 24-hour urine copper collection) may provoke excretion >1600 mcg/day in Wilson's disease, though this test is not universally accepted. Patients on treatment show markedly elevated urine copper reflecting chelator-induced excretion.
- Serum free (non-ceruloplasmin-bound) copper: Calculated as total serum copper (mcg/dL) minus (3.15 × ceruloplasmin mg/dL); values >25 mcg/dL (4.0 µmol/L) are elevated and reflect increased free copper load. Direct measurement by ultrafiltration is more accurate but less widely available.
- Serum aminotransferases: Elevated in hepatic disease; notably, in Wilsonian acute liver failure, aminotransferases may be disproportionately low (often AST/ALT <500 IU/L) because the necrotic liver has few remaining viable hepatocytes.
- Alkaline phosphatase: Characteristically low or absent in Wilsonian ALF — an important diagnostic sign that distinguishes Wilson's disease from other causes of ALF.
- Complete blood count: Hemolytic anemia (low hemoglobin, elevated reticulocytes) in ALF; thrombocytopenia from hypersplenism in cirrhosis.
- Direct Coombs test: Negative in Wilsonian hemolysis (mechanically or oxidatively mediated, not immune), distinguishing it from autoimmune hemolytic anemia.
Liver Biopsy
Liver biopsy with quantitative copper measurement is the most sensitive and specific single test for Wilson's disease. Hepatic copper content >250 mcg/g dry weight is diagnostic (normal <50 mcg/g; 50–250 mcg/g is indeterminate). The histological pattern varies with stage: early disease shows steatosis and glycogenated nuclei (hepatocytes with clear nuclei due to glycogen accumulation); progressive disease shows lobular inflammation, piecemeal necrosis, and Mallory-Denk bodies; end-stage disease shows macronodular or micronodular cirrhosis. Rhodanine stain or orcein stain can be used to visualize copper-associated protein deposits, though quantitative copper analysis is more reliable. Importantly, copper distribution is heterogeneous in cirrhosis, and a non-representative biopsy sample can yield falsely low values.
Genetic Testing
ATP7B mutation analysis by gene sequencing confirms the diagnosis and is particularly useful for family screening. However, because over 700 mutations are described and many are private (family-specific), compound heterozygosity (two different pathogenic mutations, one on each allele) is common, and negative sequencing does not fully exclude Wilson's disease. Haplotype analysis using microsatellite markers linked to the ATP7B locus is useful for presymptomatic first-degree relatives when the proband's mutations are known.
Neuroimaging
MRI of the brain is the preferred neuroimaging modality. Characteristic findings include T2-weighted hyperintensities in the putamen, caudate nucleus, thalamus, brainstem (midbrain and pons), and white matter. The "face of the giant panda" sign (T2 hyperintensity in the midbrain tegmentum with hypointense red nuclei and preserved signal in the substantia nigra) is a recognized but not pathognomonic pattern. CT may show hyperdensity in the basal ganglia from copper deposition. MRI changes often improve significantly with effective treatment.
Leipzig Scoring System
The Leipzig (Ferenci) scoring system assigns points for KF rings (2 points), neuropsychiatric symptoms (2 points), low ceruloplasmin (1–2 points), Coombs-negative hemolytic anemia (1 point), elevated 24-hour urine copper (1–2 points), high liver copper (1–2 points), and ATP7B mutation analysis (1–4 points). A total score ≥4 points strongly supports the diagnosis; 2–3 points = possible Wilson's; ≤1 point = diagnosis unlikely. This scoring system is endorsed by the European Association for the Study of the Liver (EASL) and the American Association for the Study of Liver Diseases (AASLD).
6. Treatment
Copper-Chelating Agents
D-Penicillamine (DPA) was the first effective treatment for Wilson's disease, introduced by John Walshe in 1956. It chelates copper by binding via its thiol group, promoting urinary copper excretion. Starting dose is 250 mg/day, increased gradually to 750–1500 mg/day in divided doses. Urine copper excretion rises markedly during initiation (often >2000 mcg/day in the first weeks), confirming efficacy. Maintenance dosing aims to keep 24-hour urine copper at 200–500 mcg/day.
Disadvantages of D-penicillamine include:
- Neurological paradox: Approximately 10–50% of neurological patients experience acute neurological deterioration upon starting D-penicillamine, attributed to rapid copper mobilization from the liver causing transient increase in circulating free copper; this deterioration may be irreversible in some patients
- Immunologic side effects: Drug-induced lupus erythematosus, myasthenia gravis, Goodpasture syndrome, pemphigus, nephrotic syndrome (membranous nephropathy)
- Dermatologic: Elastosis perforans serpiginosa (EPS), skin wrinkling, impaired wound healing
- Hematologic: Early leukopenia or thrombocytopenia requiring dose reduction
- Pyridoxine (vitamin B6) supplementation 25–50 mg/day is recommended as DPA inhibits pyridoxine-dependent enzymes
Trientine (triethylenetetramine) is the preferred alternative to D-penicillamine, offering similar copper chelation efficacy with a substantially better side-effect profile. Trientine chelates copper via its amino groups. Starting dose is 750–1500 mg/day in 2–3 divided doses before meals. Neurological worsening upon initiation is less common than with D-penicillamine. Trientine is first-line for patients who cannot tolerate D-penicillamine and is preferred by many experts for all presentations, particularly neurological disease. Trientine tetrahydrochloride (Cuprior) was approved by the FDA in 2022 for adult Wilson's disease patients currently treated with D-penicillamine, offering improved bioavailability.
Zinc Therapy
Zinc (as zinc acetate, zinc sulfate, or zinc gluconate) blocks intestinal absorption of copper by inducing synthesis of metallothionein in enterocytes and hepatocytes. Metallothionein binds copper with high affinity, sequestering it in enterocytes (which are then shed in feces) and limiting copper entry into the portal circulation. Zinc does not chelate and therefore does not increase urinary copper excretion.
Zinc is the preferred treatment for:
- Presymptomatic patients identified through family screening
- Maintenance therapy after initial decoppering with chelation (some centers use zinc monotherapy for maintenance)
- Pregnant women with Wilson's disease (chelators carry teratogenic risk)
- Pediatric patients with mild hepatic disease
Standard dosing: zinc acetate 50 mg elemental zinc three times daily (adults); 25 mg three times daily in children <50 kg. The main side effect is gastric irritation, reduced by taking zinc with a small amount of protein. Monitoring: 24-hour urine copper should fall below 75 mcg/day with adequate zinc therapy; 24-hour urine zinc confirms compliance (>2000 mcg/day confirms intestinal metallothionein induction).
Combination Therapy
Initial treatment of symptomatic Wilson's disease often employs a chelator (D-penicillamine or trientine) to rapidly remove accumulated copper, followed by transition to zinc monotherapy for long-term maintenance. Some centers use chelator plus zinc simultaneously, but concurrent use is generally avoided because chelators bind zinc and reduce its bioavailability; if combined, doses should be separated by at least 1 hour. EASL guidelines recommend chelation for initial treatment of symptomatic patients, with zinc for maintenance once copper is controlled (serum free copper <15 mcg/dL, 24-hour urine copper <100 mcg/day on treatment).
Liver Transplantation
Liver transplantation corrects the metabolic defect underlying Wilson's disease because the transplanted liver expresses normal ATP7B function. Transplantation is indicated for:
- Fulminant hepatic failure: Wilsonian ALF with coagulopathy and encephalopathy; mortality without transplantation approaches 100%; urgent listing is mandatory (King's score: bilirubin >10 mg/dL, INR >2, low ALP, AST, and presence of hemolysis predicts need for transplantation)
- Refractory hepatic disease: Advanced cirrhosis unresponsive to medical therapy or decompensated disease with Child-Pugh C/MELD >15
- Severe or refractory neurological disease: More controversial — transplantation is not routinely recommended for isolated neurological Wilson's disease, but may benefit patients with severe neurological impairment refractory to chelation, particularly those with underlying hepatic disease. Neurological improvement post-transplant is variable and less predictable than hepatic recovery.
Post-transplant outcomes are excellent for hepatic Wilson's disease, with 5-year survival rates of 80–90%. Copper metabolism normalizes within months post-transplant, and KF rings gradually fade.
Monitoring Treatment Response
Every 6–12 months, monitoring should include: serum ceruloplasmin, 24-hour urine copper, complete blood count, hepatic function tests, and ophthalmologic slit-lamp examination. Target serum free copper <15 mcg/dL. Patients on chelation should achieve 24-hour urine copper of 200–500 mcg/day during initial therapy, declining toward <100 mcg/day as copper depletion progresses. KF rings typically fade over 2–5 years of effective treatment. Neurological symptoms improve in most patients but may take 18–24 months; some patients plateau with residual deficits.
7. Complications
- Cirrhosis and portal hypertension: Variceal hemorrhage, ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome may supervene; management follows standard cirrhosis protocols alongside Wilson's-specific therapy
- Acute liver failure with hemolytic anemia: A medical emergency; standard ALF measures (ICU management, N-acetylcysteine, assessment for transplantation) combined with continuous venovenous hemofiltration or plasmapheresis to reduce copper load as a bridge to transplantation
- Neurological deterioration on chelation initiation: Occurs in 10–50% of neurological patients starting D-penicillamine; partially mitigated by using trientine or starting at very low doses; may require temporary dose reduction
- Renal Fanconi syndrome: Proximal tubular dysfunction causing electrolyte wasting; monitor serum phosphate, uric acid, and bicarbonate; supplement as needed
- Osteoporosis: Calcium and vitamin D supplementation; DEXA scanning; bisphosphonates if clinically indicated
- Drug side effects: D-penicillamine — drug-induced lupus, nephrotic syndrome, myasthenia gravis requiring drug discontinuation and switch to trientine; zinc — gastric irritation (manageable by formulation change)
- Treatment non-adherence: A major cause of relapse and acute decompensation; patients must understand that treatment is lifelong; even brief cessation (weeks to months) can precipitate fulminant ALF in previously stable patients
- Hepatocellular carcinoma (HCC): Risk is significantly lower in Wilson's disease cirrhosis than in viral or alcoholic cirrhosis, but HCC screening is still recommended by AASLD guidelines in cirrhotic patients (ultrasound every 6 months)
- Featured Videos
8. Prognosis
Wilson's disease is uniformly fatal without treatment. With appropriate copper-lowering therapy started before advanced organ damage, prognosis is excellent — most patients achieve near-normal life expectancy and quality of life. Key prognostic factors include:
- Presentation phenotype: Hepatic disease (excluding fulminant ALF) generally responds well to chelation; neurological disease also improves but recovery is slower and less complete
- Stage at diagnosis: Pre-cirrhotic or compensated cirrhotic patients respond better than those with decompensated cirrhosis or fulminant ALF
- Neurological severity: Patients with dysarthria and tremor as predominant features respond better than those with severe dystonia or dementia; MRI brain lesions often improve with treatment
- Treatment adherence: The strongest modifiable prognostic factor; treatment cessation at any stage carries risk of acute decompensation and death
- Pregnancy: Wilson's disease does not preclude pregnancy; zinc is the preferred agent; chelation doses may need adjustment as ceruloplasmin rises physiologically in pregnancy; untreated Wilson's disease is associated with recurrent pregnancy loss
Patients with Wilsonian acute liver failure who receive transplantation have 1-year post-transplant survival rates of 75–80%. Patients with purely neurological Wilson's disease who are diagnosed early and treated consistently may have complete or near-complete neurological recovery, particularly with trientine-based regimens.
9. Prevention and Screening
Because Wilson's disease is autosomal recessive, each first-degree sibling of an affected individual has a 1 in 4 risk of being affected and a 1 in 2 risk of being a carrier. Screening of all first-degree siblings is mandatory once a proband is identified. The recommended screening protocol includes:
- Serum ceruloplasmin and 24-hour urine copper
- Slit-lamp examination for Kayser-Fleischer rings
- Liver function tests and complete blood count
- ATP7B haplotype analysis or mutation sequencing (if the proband's mutations are known)
Presymptomatic individuals diagnosed through family screening should be treated with zinc (the preferred agent given its safety profile) to prevent copper accumulation and organ damage. Early treatment of presymptomatic disease is highly effective, and treated presymptomatic patients typically remain disease-free indefinitely.
Neonatal or population-based screening for Wilson's disease is not currently feasible due to the absence of a reliable newborn biomarker and the late onset of symptoms; however, next-generation sequencing-based newborn screening panels are under investigation. Dietary copper restriction (avoidance of shellfish, liver, chocolate, mushrooms, and nuts in large quantities) is a sensible adjunct to medical therapy but cannot replace pharmacological copper removal.
10. Recent Research and Advances
- Gene therapy: Adeno-associated virus (AAV) vectors delivering wild-type ATP7B to hepatocytes have shown promising results in animal models, correcting copper metabolism and reversing liver disease. Phase 1/2 clinical trials (e.g., VTX-801 / Aavid Therapeutics) have been initiated in adults with Wilson's disease; early results suggest safety and potential efficacy in reducing copper burden. Gene therapy may eventually provide a one-time cure, eliminating the need for lifelong pharmacotherapy.
- Trientine tetrahydrochloride (TETA-4HCl / Cuprior): FDA-approved in 2022 for Wilson's disease patients currently treated with D-penicillamine who require a change in therapy; the tetrahydrochloride salt formulation provides more predictable oral bioavailability than trientine dihydrochloride and is stable at room temperature (overcoming the cold-chain requirement of older trientine formulations).
- Molecular mechanisms of ATP7B trafficking: Research into how ATP7B vesicular trafficking is regulated under varying copper loads — and how missense mutations disrupt protein folding vs. trafficking vs. catalytic activity — has opened pharmacochaperone approaches: small molecules that rescue misfolded but partially functional ATP7B variants from proteasomal degradation, similar to CFTR corrector/potentiator strategies in cystic fibrosis.
- Biomarkers for treatment monitoring: Direct measurement of non-ceruloplasmin-bound copper (NCC) by ultrafiltration with graphite furnace atomic absorption spectrometry provides a more accurate reflection of copper toxicity than calculated free copper; NCC >1.6 µmol/L correlates with disease activity and may supplant traditional monitoring parameters.
- Neuroimaging outcome data: Long-term MRI studies demonstrate significant improvement in basal ganglia lesions and white matter hyperintensities in patients treated effectively with trientine compared to D-penicillamine, supporting trientine preference for neurological Wilson's disease.
- Wilson Index / revised scoring: Refinements to the Wilsonian ALF King's score (incorporating leukocyte count and the alkaline phosphatase-to-bilirubin ratio) are being validated to more accurately identify patients who will not survive without liver transplantation, enabling earlier and more decisive transplant listing decisions.
11. References
- Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson's disease. Lancet. 2007;369(9559):397–408 — Search PubMed
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089–2111 — Search PubMed
- European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson's disease. Journal of Hepatology. 2012;56(3):671–685 — Search PubMed
- Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver International. 2003;23(3):139–142. PMID: 12955875
- Członkowska A, Litwin T, Dusek P, et al. Wilson disease. Nature Reviews Disease Primers. 2018;4(1):21 — Search PubMed
- Walshe JM. Wilson's disease: the importance of measuring serum caeruloplasmin non-immunologically. Annals of Clinical Biochemistry. 2003;40(Pt 2):115–121 — Search PubMed
- Brewer GJ, Askari F, Lorincz MT, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Archives of Neurology. 2006;63(4):521–527. PMID: 16606763
- Weiss KH, Thurik IF, Gotthardt DN, et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease: a prospective, open-label, multicenter, cohort study (EUROWILSON). Orphanet Journal of Rare Diseases. 2013;8:78 — Search PubMed
- Schilsky ML. Wilson disease: diagnosis, treatment, and follow-up. Clinics in Liver Disease. 2017;21(4):755–767. PMID: 28987255
- Catana AM, Medici V. Liver transplantation for Wilson disease. World Journal of Hepatology. 2012;4(1):5–10 — Search PubMed
- Linn FH, Houwen RH, van Hattum J, van der Kleij SF, van Erpecum KJ. Long-term exclusive zinc monotherapy in symptomatic Wilson disease: experience in 17 patients. Hepatology. 2009;50(5):1442–1452 — Search PubMed
- Coffey AJ, Durkie M, Hague S, et al. A genetic study of Wilson's disease in the United Kingdom. Brain. 2013;136(Pt 5):1476–1487. PMID: 23518715
Research Papers
Explore current literature on Wilson's disease via PubMed topic searches. These links open live PubMed searches for the listed keywords — results update as new studies are indexed.
- Wilson's disease ATP7B genetics — PubMed search
- Wilson's disease ceruloplasmin diagnosis — PubMed search
- Wilson's disease trientine chelation — PubMed search
- Wilson's disease D-penicillamine — PubMed search
- Wilson's disease zinc maintenance therapy — PubMed search
- Wilson's disease liver transplantation — PubMed search
- Wilson's disease gene therapy — PubMed search
- Kayser-Fleischer rings copper cornea — PubMed search
Connections
- Nephrology & Hepatology
- Cirrhosis
- Liver Disease
- Non-Alcoholic Fatty Liver Disease
- Hepatic Encephalopathy
- Hemochromatosis
- Hepatitis
- Acute Kidney Injury
- Zinc
- Copper
- Neurology Overview
- Ceruloplasmin
- Primary Biliary Cholangitis
- Copper Toxicity and Wilson's Disease — the wider picture of copper overload, acute and chronic.
- Copper Toxicity: Symptoms and Risks — what excess copper does to the liver, brain, and red blood cells.
- Copper Toxicity: Neurological and Psychiatric Symptoms — the tremor, dystonia, and mood changes that copper in the brain produces.
- Copper Test (Serum Copper and Ceruloplasmin) — how the copper panel is read, and why serum copper alone misleads here.
- Copper Toxicity: Nausea and Stomach Upset — the acute gastric reaction to swallowed copper, contrasted with this slow overload.