Iodine for Thyroid Function
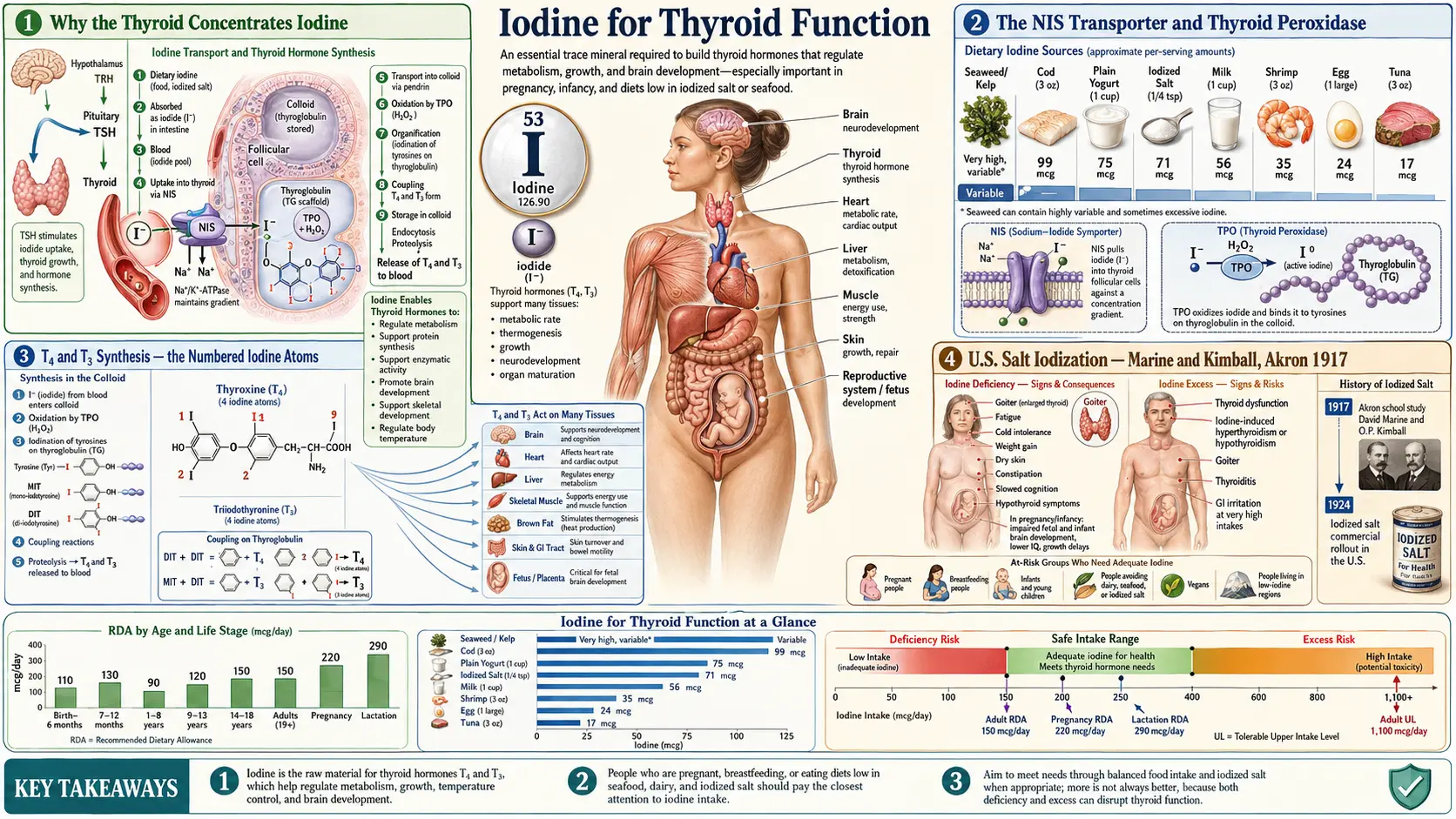
The thyroid gland is the dominant iodine-consuming organ in the body, and the synthesis of thyroxine (T4, four iodine atoms) and triiodothyronine (T3, three iodine atoms) is the single most important biochemical use of iodine that exists. Without adequate iodide arriving at the sodium-iodide symporter (NIS) on the basolateral membrane of the thyroid follicular cell, the entire downstream cascade — thyroid peroxidase (TPO) iodination of tyrosine, coupling into iodothyronines, storage in thyroglobulin colloid, release into the bloodstream, peripheral conversion to active T3, and binding to nuclear thyroid hormone receptors in every tissue — collapses. This page walks through that cascade in clinical detail, the historical achievement of universal salt iodization (Marine and Kimball in Akron, Ohio, 1917), the Wolff-Chaikoff effect and its escape, the modern high-dose iodine protocols of David Brownstein and Guy Abraham, the pharmacology of Lugol's solution, and the iodide-vs-molecular-iodine question that determines which form goes to which tissue.
Table of Contents
- Why the Thyroid Concentrates Iodine
- The NIS Transporter and Thyroid Peroxidase
- T4 and T3 Synthesis — the Numbered Iodine Atoms
- U.S. Salt Iodization — Marine and Kimball, Akron 1917
- The Wolff-Chaikoff Effect and Escape
- Iodine Deficiency, Goiter, and Hypothyroidism
- Brownstein and Abraham High-Dose Iodine Protocols
- Lugol's Solution and Iodide-vs-Molecular-Iodine
- Selenium — the Required Cofactor
- Hashimoto's and the Iodine Controversy
- Key Research Papers
- Connections
- Featured Videos
Why the Thyroid Concentrates Iodine
The human thyroid gland weighs only 15-25 grams in an adult but contains approximately 70-80% of the body's total iodine pool — about 15-20 milligrams of iodine in an iodine-replete adult. This concentration ratio is extraordinary: the thyroid contains roughly 1,500 times more iodine per gram of tissue than the surrounding blood plasma. The reason is biochemical necessity. Thyroid hormones are unique in human biology because they contain a halogen (iodine) at four specific positions on a coupled-tyrosine backbone, and there is no synthesis pathway that does not require dietary iodine as the input atom.
Plant and bacterial biochemistry has dozens of metabolic alternatives. Human thyroid hormone synthesis has one. Iodine deficiency therefore produces a deficiency that cannot be substituted from any other dietary input, and the resulting clinical syndrome — hypothyroidism, goiter, cretinism, and the full spectrum of "iodine deficiency disorders" (IDD) as Basil Hetzel of the WHO named them in the 1980s — is one of the few nutritional deficiency syndromes that has a single specific cure.
The evolutionary logic is interesting. Iodine is abundant in seawater (about 50 µg/L) and accumulates dramatically in marine algae (kelp can reach 1,500-8,000 ppm dry weight). Early human populations living on coasts ingested several thousand micrograms of iodine per day from seaweed and seafood. As populations migrated inland and adopted agricultural diets dependent on rain-leached, iodine-poor soils, the average intake fell to a few tens of micrograms per day — far below the 150 µg/day required to prevent goiter. The "goiter belts" of the world — the U.S. Great Lakes region, Switzerland, the Himalayan foothills, the Andes, much of inland China — map almost perfectly onto geological regions with iodine-depleted soils.
The NIS Transporter and Thyroid Peroxidase
The sodium-iodide symporter (NIS) is a 13-pass transmembrane glycoprotein encoded by the SLC5A5 gene, expressed on the basolateral membrane of thyroid follicular cells. It is the molecular gate through which all iodide entering the thyroid must pass. NIS uses the sodium gradient maintained by Na+/K+ ATPase to co-transport iodide against its concentration gradient, achieving a thyroid-to-plasma iodide ratio of 20-40 in the basal state, and substantially higher under thyroid-stimulating hormone (TSH) stimulation.
Several environmental anions compete with iodide for NIS uptake. The order of competitive affinity is approximately: perchlorate > thiocyanate > iodide > nitrate. This is clinically important because:
- Perchlorate (ClO4⁻) — a NASA rocket-fuel oxidizer, a contaminant of many groundwater supplies in the western United States, and used pharmaceutically (potassium perchlorate) to block thyroid uptake of iodide in iodine-induced hyperthyroidism
- Thiocyanate (SCN⁻) — the principal goitrogen in cigarette smoke (from hydrogen cyanide metabolism) and in cruciferous vegetables (from glucosinolate hydrolysis), explaining why smokers and high-cruciferous-vegetable consumers have higher iodine requirements
- Nitrate (NO3⁻) — a competitor at high concentrations, relevant to agricultural runoff contamination of drinking water
Once inside the follicular cell, iodide is transported across the apical membrane into the colloid lumen by the pendrin transporter (encoded by SLC26A4; mutations cause Pendred syndrome, a combined deafness-goiter syndrome). At the apical surface, thyroid peroxidase (TPO) — a heme-containing enzyme — oxidizes iodide to a reactive iodinating species (iodinium ion or iodine radical) using hydrogen peroxide (H2O2) generated by the dual oxidase enzymes DUOX1 and DUOX2. The activated iodine then attaches to specific tyrosine residues on the giant 660 kDa thyroglobulin glycoprotein stored in the colloid.
T4 and T3 Synthesis — the Numbered Iodine Atoms
The thyroid hormone names are not arbitrary — they describe the molecular structure with precision. Thyroxine (T4) contains four iodine atoms; triiodothyronine (T3) contains three. The "T" stands for "thyronine," the coupled-tyrosine backbone. The "4" or "3" is literally the count of iodine atoms attached to that backbone at the 3, 5, 3', and 5' positions of the two aromatic rings.
The synthesis sequence on thyroglobulin tyrosine residues is:
- A single tyrosine residue gets one iodine attached → monoiodotyrosine (MIT) — one iodine atom
- A different tyrosine residue gets two iodines attached → diiodotyrosine (DIT) — two iodine atoms
- Two adjacent DIT residues couple (TPO-catalyzed phenol coupling) → thyroxine (T4) — 2 + 2 = four iodine atoms
- A DIT plus an MIT couple → triiodothyronine (T3) — 2 + 1 = three iodine atoms
The thyroid produces about 80% T4 and 20% T3 directly. T4 is the major circulating form (half-life ~7 days) and functions essentially as a prohormone. The biological activity is in T3, which is generated peripherally by deiodinase enzymes that remove one iodine from T4. The 5'-deiodinase (type 1 and type 2, both selenium-dependent) converts T4 to active T3 by removing the outer-ring iodine; the type 3 deiodinase removes the inner-ring iodine to generate reverse T3 (rT3), the inactive isomer that competitively occupies T3 receptors without activating them.
This synthesis architecture has clinical implications. Because T4 carries 4 iodine atoms and T3 carries only 3, the thyroid under iodine restriction will preferentially synthesize T3 to conserve iodine — an adaptive response visible as a rising T3/T4 ratio on labs in early-to-moderate iodine deficiency, before frank hypothyroidism appears.
U.S. Salt Iodization — Marine and Kimball, Akron 1917
The history of iodine prophylaxis in the United States is one of public health's most successful and least-remembered interventions. In 1917, David Marine (a Cleveland pathologist) and Oliver Kimball persuaded Akron, Ohio school authorities to enroll roughly 4,500 schoolgirls in a controlled trial of sodium iodide prophylaxis (2 grams sodium iodide given over 10 days, twice per year). The schoolgirls were chosen because adolescent girls in the iodine-poor Great Lakes "goiter belt" had a goiter prevalence of 50% or higher, with the visible thyroid enlargement often progressing through puberty.
The results, published in 1920 in the Journal of the American Medical Association, were dramatic. Goiter prevalence in the treated group fell from baseline to almost zero. Goiter incidence in the untreated control group continued at the baseline rate. The trial was the first definitive proof that iodine deficiency caused goiter and that simple oral prophylaxis prevented it.
In 1924, Morton Salt introduced commercially iodized table salt (potassium iodide added at approximately 76 ppm iodine, equivalent to 76 mg per kg salt; a teaspoon contains about 400 mcg of iodine). The intervention was voluntary and was adopted region-by-region over the following decade. By the 1950s, U.S. goiter prevalence had fallen by more than 90% in formerly endemic areas. The intervention has since been adopted by most countries through the WHO and Iodine Global Network's Universal Salt Iodization (USI) program, which by 2020 covered approximately 88% of households worldwide.
The complication for the modern era is that:
- About 70% of U.S. sodium intake now comes from processed and restaurant foods, which almost universally use non-iodized salt
- Sodium-restriction public health messaging has reduced the use of table salt at home
- The commercial baking industry switched from potassium iodate dough conditioners to potassium bromate in the 1960s (replacing an iodine source with a bromine source that actively competes with iodine for NIS uptake)
- Median urinary iodine in the U.S. NHANES dataset fell from about 320 µg/L in 1971-1974 to about 145 µg/L in 2001-2002, and has hovered just above the WHO sufficiency threshold (100 µg/L) ever since
For the relevant deeper background on the modern environmental halide load, see our Fluoride page. Fluoride is another halide competitor for thyroid function and a separate component of the modern iodine-displacement burden.
The Wolff-Chaikoff Effect and Escape
The Wolff-Chaikoff effect is the acute, transient suppression of thyroid hormone synthesis that occurs when the thyroid is suddenly exposed to a large iodide load (typically > 1-2 mg in a few hours). Discovered by Jan Wolff and Israel Lyon Chaikoff at UC Berkeley in 1948, the mechanism is a feedback inhibition of TPO by high intra-thyroidal iodide concentration — the thyroid temporarily downregulates iodination to prevent runaway hormone synthesis in an iodine-rich environment.
The Wolff-Chaikoff effect was originally interpreted as evidence that high-dose iodine is dangerous because it shuts off thyroid hormone production. What was missed for several decades is the second half of the story: escape from the Wolff-Chaikoff effect. In the normal thyroid, the acute suppression lasts only about 24-48 hours. The gland then "escapes" by downregulating NIS expression on its basolateral membrane, reducing iodide uptake to levels at which TPO can resume normal synthesis. The thyroid achieves a new steady state in which it is consuming the necessary iodide for hormone synthesis while keeping intracellular iodide concentration below the level that inhibits TPO.
Clinical implications:
- In the normal thyroid, the Wolff-Chaikoff suppression is transient (24-48 hours) and clinically inconsequential at any iodine intake compatible with life
- In thyroid pathologies with impaired NIS regulation — particularly autoimmune thyroiditis (Hashimoto's) where the regulatory machinery may be damaged — the escape can fail, producing iodine-induced hypothyroidism with sustained high-dose exposure
- In thyroid pathologies with autonomous nodules or Graves' disease, a high-dose iodine bolus can instead produce Jod-Basedow phenomenon — iodine-induced hyperthyroidism — because the autonomous nodule lacks the normal feedback regulation that would otherwise downregulate NIS
- The Wolff-Chaikoff effect is the explicit pharmacologic basis for the use of Lugol's solution or saturated solution of potassium iodide (SSKI) in preparing a patient for thyroidectomy — the gland is intentionally suppressed for 10-14 days before surgery to reduce vascularity and ease the operation
Iodine Deficiency, Goiter, and Hypothyroidism
The clinical sequence of iodine deficiency is well-characterized. Initially the thyroid compensates by upregulating NIS expression, increasing its iodine-trapping efficiency, and shifting hormone production toward T3 (which requires one fewer iodine atom than T4). Serum TSH rises as the pituitary senses falling T4. Under sustained TSH stimulation, the thyroid hypertrophies — the follicular cells enlarge, multiply, and increase colloid storage capacity, producing visible gland enlargement (goiter).
Goiter is the classic clinical sign of iodine deficiency, visible as a swelling at the front of the neck centered on the cricoid cartilage. In children and adolescents, the goiter is typically diffuse and smooth. In adults with long-standing deficiency, the gland often becomes nodular — multinodular goiter — with discrete nodules that may become functionally autonomous (producing thyroid hormone independent of TSH regulation, a setup for iodine-induced hyperthyroidism if iodine intake suddenly increases).
Hypothyroidism develops when the thyroid's compensatory hypertrophy can no longer keep up with the demand for hormone production. Clinical features include:
- Fatigue, cold intolerance, sluggish thinking, depressed mood
- Constipation, dry skin, brittle hair, thinning of the outer third of the eyebrows ("Queen Anne sign")
- Weight gain (modest — usually 5-10 lbs, mostly fluid; severe weight gain is rarely attributable to hypothyroidism alone)
- Bradycardia, diastolic hypertension, elevated LDL cholesterol
- Menstrual irregularities (heavy periods, infertility, miscarriage)
- In severe long-standing untreated cases, myxedema — the puffy, doughy skin and tongue thickening from glycosaminoglycan accumulation
For the iodine-related downstream conditions, see our pages on Hashimoto's Thyroiditis, Thyroid Disorders, and the related Metabolism & Energy deep-dive that addresses the clinical pattern of stuck-at-high-weight patients who diet without losing.
Brownstein and Abraham High-Dose Iodine Protocols
The mainstream iodine recommendation in the U.S. is the Institute of Medicine RDA of 150 µg/day for adults (220 µg/day in pregnancy, 290 µg/day in lactation), and the WHO sufficiency target of urinary iodine ≥ 100 µg/L. These targets were set specifically to prevent goiter and cretinism — the most severe end of the iodine-deficiency-disorder spectrum — and were calibrated to the iodine intake at which thyroid hormone production reaches steady state.
Beginning in the late 1990s, a distinct clinical school led by Guy Abraham (a former Harvard medical school faculty endocrinologist) and Jorge Flechas, and popularized clinically by David Brownstein, argued that the RDA is far too low and represents the minimum to prevent visible thyroid pathology, not the optimum to saturate all iodine-utilizing tissues in the body. The argument is grounded in:
- Extra-thyroidal NIS expression — the breast, salivary glands, gastric mucosa, ovaries, prostate, lacrimal glands, and choroid plexus all concentrate iodine. None of these tissues was considered when the original goiter-prevention RDA was set.
- Japanese dietary iodine intake — the average Japanese person consumes roughly 1,000-3,000 µg of iodine per day from seaweed and seafood, with no apparent harm and historically low rates of breast, prostate, and thyroid cancer.
- Iodine loading test — Abraham introduced a clinical test in which a 50 mg oral dose of iodine/iodide is given and 24-hour urinary iodine is measured. In iodine-sufficient subjects, > 90% of the dose is excreted within 24 hours; in iodine-deficient subjects, a large fraction is retained as the tissues take up the load.
- Bromide and halide displacement — high-dose iodine appears to displace bromide and fluoride from tissue stores, with measurable increases in urinary bromide excretion over the weeks following supplementation initiation.
The Brownstein protocol typically uses 12.5-50 mg of iodine (combined iodide plus molecular iodine, as in Iodoral tablets or Lugol's solution drops) per day for adult repletion, with selenium (200-400 mcg), magnesium, vitamin C, and unrefined salt as obligate cofactors. The thyroid is monitored with TSH, free T4, free T3, TPO antibodies, and thyroglobulin antibodies at baseline and at 3-6 month intervals.
The clinical safety record of these protocols, as reported by Brownstein and colleagues in case series, has been favorable when the cofactor support is rigorous. The mainstream endocrinology community remains skeptical, primarily citing:
- The risk of iodine-induced hyperthyroidism (Jod-Basedow) in patients with autonomous nodules
- The risk of triggering or worsening Hashimoto's thyroiditis in genetically susceptible individuals (though Brownstein argues this is iatrogenic from inadequate selenium support)
- The lack of large prospective randomized trials
The pragmatic clinical position, supported by integrative practitioners with substantial experience, is that high-dose iodine is reasonable in selected patients with documented insufficiency or specific clinical indications (fibrocystic breast disease, recurrent ovarian cysts, prostate enlargement), with appropriate cofactor support and lab monitoring, and with conservative initial dosing (start at 3-6 mg/day and titrate up over weeks).
Lugol's Solution and Iodide-vs-Molecular-Iodine
Lugol's solution is a 5% iodine plus 10% potassium iodide solution in water, formulated by the French physician Jean Lugol in 1829. The original 5% formulation contains approximately 6.25 mg of total iodine per drop (about 50 mg/mL). A 2% Lugol's solution contains 2.5 mg per drop, and various reformulations exist. The dual iodine-plus-iodide composition is biochemically deliberate:
- Iodide (I⁻) — the form preferentially concentrated by the thyroid (and salivary, gastric, lactating-breast NIS expression). The thyroid's native substrate for hormone synthesis.
- Molecular iodine (I₂) — the form preferentially concentrated by the non-lactating breast and prostate, and the form responsible for the apoptosis-promoting and antioxidant effects in non-thyroid tissues. The Aceves group at UNAM Mexico has shown that I₂ specifically, not iodide, drives the iodolactone formation that activates PPAR-γ and promotes apoptosis in breast cancer cell lines.
This pharmacology has implications for what to take. Pure potassium iodide (KI) tablets — the form distributed for nuclear radiation prophylaxis — deliver iodide only and may be suboptimal for the extra-thyroidal tissues. Lugol's and Iodoral (the tableted form of Lugol's, 12.5 mg per tablet, 7.5 mg potassium iodide plus 5 mg molecular iodine) deliver both forms. Sea-vegetable iodine (kelp, dulse, nori) delivers a mix that is mostly iodide.
The clinical signature of low-quality iodine supplementation: a patient on KI alone for fibrocystic breast disease may experience less benefit than expected, because the molecular iodine that the breast preferentially uses is not in the supplement.
Selenium — the Required Cofactor
Selenium and iodine cannot be considered separately in thyroid biology. The thyroid uses hydrogen peroxide (H2O2) generated by DUOX enzymes as the oxidant required by TPO to iodinate tyrosine residues. Excess H2O2 not consumed by TPO is potentially cytotoxic. The glutathione peroxidase family (GPx1, GPx3, and notably the selenoprotein iodothyronine deiodinases) provides the antioxidant defense that protects the thyroid from this self-generated oxidative stress. All of these enzymes require selenium as the active-site selenocysteine residue.
In selenium-replete individuals, TPO operates at full capacity and the H2O2 byproduct is safely neutralized. In selenium-deficient individuals, H2O2 accumulates, damages the thyroid follicular cell membrane, and exposes intracellular antigens (notably TPO itself and thyroglobulin) to circulating lymphocytes — potentially triggering or accelerating Hashimoto's thyroiditis.
This is the mechanistic basis for the integrative-medicine rule: do not supplement iodine without ensuring adequate selenium. Recommendations vary slightly across practitioners, but a typical protocol pairs:
- 200-400 mcg/day selenomethionine (or a mixed selenium product)
- One Brazil nut per day provides approximately 70-90 mcg of selenium and is a reasonable food source for those who prefer dietary repletion
- Selenium should be in place for at least 2-4 weeks before high-dose iodine is introduced
For more on selenium's role in thyroid biology and the broader thyroid-supportive nutrient stack, see our Selenium page.
Hashimoto's and the Iodine Controversy
Hashimoto's thyroiditis is the autoimmune destruction of the thyroid gland mediated by anti-TPO and anti-thyroglobulin antibodies. It is the most common cause of hypothyroidism in iodine-sufficient countries and its prevalence has been rising for decades. The conventional position is that iodine supplementation is contraindicated in Hashimoto's based on epidemiologic observations that aggressive iodine fortification programs in formerly deficient populations (notably China and Sri Lanka in the 1990s) have been followed by measurable increases in thyroid autoimmunity.
The integrative counter-position, advanced by Brownstein, Flechas, Abraham, and others, is that the observed autoimmunity increase is not caused by iodine itself but by the failure to provide adequate selenium cofactor at the same time. The mechanistic argument is that suddenly increasing iodine in a selenium-deficient population overwhelms TPO with H2O2 it cannot safely neutralize, damages thyroid cells, and exposes the autoantigens that drive the autoimmune cascade.
The middle-ground clinical position that has emerged:
- For patients with established Hashimoto's (positive TPO or thyroglobulin antibodies), start with selenium repletion alone (200-400 mcg/day) for 6-12 weeks and recheck antibodies
- Selenium alone produces a measurable reduction in TPO antibody titers in most patients (multiple randomized trials confirm this; the Gartner 2002 trial is the seminal reference)
- If iodine is to be introduced, start very low (a few hundred mcg/day, not the 12.5-50 mg of the high-dose Brownstein protocol) and titrate slowly
- Monitor TSH, free T4, free T3, TPO antibodies, and thyroglobulin antibodies at 3-month intervals during titration
- If antibody titers rise on iodine, back off; if they fall or remain stable, continue
- Patients with documented frank iodine deficiency by urinary iodine loading test may benefit more clearly than patients with normal baseline iodine status
Key Research Papers
- Marine D, Kimball OP (1920). Prevention of simple goiter in man. JAMA. — PubMed
- Wolff J, Chaikoff IL (1948). Plasma inorganic iodide as a homeostatic regulator of thyroid function. JBC. — PubMed
- Dai G, Levy O, Carrasco N (1996). Cloning and characterization of the thyroid iodide transporter. Nature. — PubMed
- Hetzel BS (1983). Iodine deficiency disorders (IDD) and their eradication. Lancet. — PubMed
- Abraham GE (2005). The historical background of the iodine project. The Original Internist. — PubMed
- Brownstein D, clinical experience with high-dose iodine supplementation — PubMed
- Gartner R et al. (2002). Selenium supplementation in patients with autoimmune thyroiditis decreases thyroid peroxidase antibody concentrations. JCEM. — PubMed
- Zimmermann MB (2009). Iodine deficiency. Endocrine Reviews. — PubMed
- Pearce EN et al. (2013). Sources of dietary iodine: bread, milk, and fish. Best Pract Res Clin Endocrinol Metab. — PubMed
- Caldwell KL et al. (2011). Iodine status of the U.S. population, NHANES 2005-2008. Thyroid. — PubMed
- Markou K et al. (2001). Iodine-induced hypothyroidism. Thyroid. — PubMed
- Vagenakis AG, Braverman LE (1975). Adverse effects of iodides on thyroid function. Med Clin North Am. — PubMed
PubMed Topic Searches
- PubMed: NIS sodium-iodide symporter
- PubMed: TPO iodination mechanism
- PubMed: Wolff-Chaikoff escape
- PubMed: Universal salt iodization
- PubMed: Selenium and Hashimoto's
Connections
- Iodine Benefits Hub
- Iodine Overview
- Iodine for Brain Development
- Iodine for Breast Health
- Iodine for Metabolism & Energy
- Selenium (TPO cofactor)
- Tyrosine (T4/T3 backbone)
- Zinc
- Hashimoto's Thyroiditis
- Thyroid Disorders
- Thyroid Cancer
- Fluoride (NIS competitor)
- Heavy Metals
- Fatigue
- All Minerals