Histidine, Histamine, and Allergy
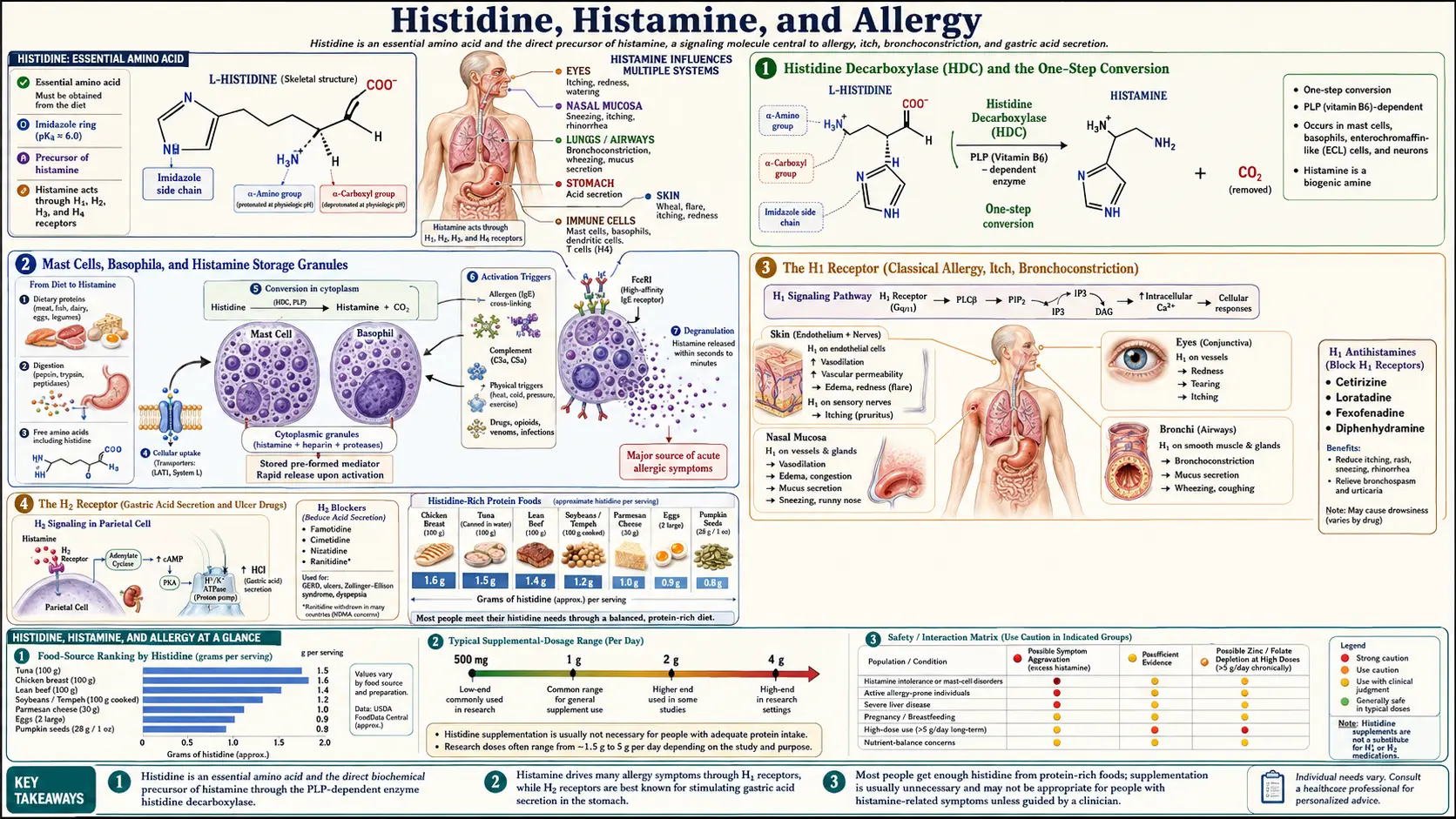
Histidine is the only proteinogenic amino acid that the body converts directly into a neurotransmitter and inflammatory mediator at industrial scale — a single enzyme, histidine decarboxylase (HDC), strips off the carboxyl group and produces histamine. Histamine then signals through four distinct G-protein-coupled receptors (H1, H2, H3, H4) that span allergy, gastric acid secretion, central nervous system arousal, and immune cell recruitment. This is also the deepest pharmacology rabbit hole in the amino acid kingdom: the first-generation antihistamines (Benadryl) and the second-generation non-sedating antihistamines (Claritin, Allegra, Zyrtec) all block H1; the H2 blockers (cimetidine, ranitidine, famotidine) revolutionized peptic ulcer disease in the 1970s and earned a Nobel-adjacent legacy; the H3 receptor is a hot target for narcolepsy and ADHD drug development; and the H4 receptor is the new frontier in chronic pruritus and inflammatory bowel disease. This deep-dive walks through histidine's journey to histamine, the receptor pharmacology that powers a multi-billion-dollar drug class, mast cell activation syndrome (MCAS), the low-histamine diet for chronic urticaria, and the DAO/HNMT degradation enzymes that determine whether ingested histamine causes you symptoms or passes through unnoticed.
Table of Contents
- Histidine Decarboxylase (HDC) and the One-Step Conversion
- Mast Cells, Basophils, and Histamine Storage Granules
- The H1 Receptor (Classical Allergy, Itch, Bronchoconstriction)
- The H2 Receptor (Gastric Acid Secretion and Ulcer Drugs)
- The H3 Autoreceptor (CNS Arousal, Narcolepsy, ADHD)
- The H4 Receptor (Pruritus, Chronic Inflammation, IBD)
- DAO and HNMT: The Two Histamine Degradation Enzymes
- Mast Cell Activation Syndrome (MCAS)
- The Low-Histamine Diet for Chronic Urticaria and Intolerance
- A Brief History of the Antihistamine Drug Class
- Key Research Papers
- Connections
- Featured Videos
Histidine Decarboxylase (HDC) and the One-Step Conversion
Of all 20 proteinogenic amino acids, only a small handful are precursors to monoamine signaling molecules: tryptophan to serotonin and melatonin, tyrosine to dopamine and the catecholamines, glutamate to GABA, and histidine to histamine. The histidine-to-histamine conversion is the simplest of all these pathways — a single enzymatic step performed by histidine decarboxylase (HDC), a pyridoxal-5'-phosphate (vitamin B6 active form) dependent enzyme that removes the alpha-carboxyl group of histidine and releases CO2, leaving behind histamine.
The reaction is so simple that it defines a metabolic bottleneck: the rate of histamine production in any tissue is determined almost entirely by HDC enzyme expression and by the local availability of histidine and B6 cofactor. HDC is expressed at high levels in only a few specialized cell types — mast cells throughout connective tissue and at mucosal surfaces, basophils circulating in peripheral blood, enterochromaffin-like (ECL) cells in the gastric mucosa, and a small population of histaminergic neurons clustered exclusively in the tuberomammillary nucleus of the hypothalamus. This restricted expression pattern is why histamine signaling is so anatomically specific and so therapeutically targetable.
HDC activity is upregulated by inflammation, IgE crosslinking on mast cells, gastrin in the stomach, and a circadian rhythm in the brain. It is downregulated by glucocorticoids (one of several mechanisms behind their anti-inflammatory action) and by alpha-methylhistidine, a research-tool HDC inhibitor that has been used to map the contribution of newly-synthesized versus pre-stored histamine to various physiological responses.
Practical implication: because the conversion requires vitamin B6 as a cofactor, marginal B6 deficiency can subtly reduce histamine production capacity. This rarely produces frank symptoms in healthy adults but can contribute to suboptimal immune readiness and may interact with the carbidopa/levodopa pharmacology in Parkinson's patients (where the same B6 cofactor is needed for the closely-related DOPA decarboxylase).
Mast Cells, Basophils, and Histamine Storage Granules
Once synthesized, histamine is packaged into membrane-bound secretory granules within mast cells and basophils. Each mast cell contains hundreds to thousands of granules, and each granule contains histamine alongside other pre-formed mediators including heparin, tryptase, chymase, and a cocktail of cytokines and chemokines. The storage form is highly stable — histamine binds to the negatively-charged heparin proteoglycan matrix within the granule and is held essentially inert until release.
Mast cells are positioned at strategic sites where the body interfaces with the outside world — just beneath the skin epidermis, lining the respiratory and gastrointestinal mucosa, perivascular in connective tissue, and abundantly in the brain meninges. They function as sentinel cells that detect parasites, allergens, and certain bacterial products, and that trigger the local inflammatory response when needed. When activated, mast cells undergo degranulation, releasing the entire granule contents into the extracellular space within seconds.
Activation can be triggered by several distinct mechanisms:
- IgE crosslinking — the classical allergy pathway. Allergen-specific IgE antibodies bind to high-affinity Fc-epsilon-RI receptors on the mast cell surface. When allergen binds and crosslinks adjacent IgE molecules, the receptor aggregation triggers degranulation within minutes. This is the mechanism behind hay fever, food allergy, and anaphylaxis.
- Complement anaphylatoxins (C3a, C5a) — generated during innate immune complement activation, these peptides directly stimulate mast cell degranulation independent of IgE.
- Substance P and other neuropeptides — the neuroimmune connection. Sensory nerves release substance P during tissue stress, which directly activates mast cells. This is one mechanism behind neurogenic inflammation and the rosacea-stress connection.
- MRGPRX2 receptor activation — a non-IgE pathway discovered more recently. Many drugs (opioids, vancomycin, neuromuscular blockers, fluoroquinolones), insect venoms, and certain endogenous peptides activate this receptor and cause pseudo-allergic reactions that look like anaphylaxis but lack the IgE component.
- Mechanical pressure and temperature — the basis for dermographism (mast cells release histamine in response to scratching) and cold urticaria.
The dual-pathway architecture (IgE-mediated versus MRGPRX2-mediated activation) explains why some patients with negative allergy testing still get hives and flushing from certain drugs — their reactions are pseudoallergic, mast-cell-mediated but not IgE-driven, and conventional allergy testing misses them entirely.
The H1 Receptor (Classical Allergy, Itch, Bronchoconstriction)
Once released, histamine signals through four distinct receptor subtypes, each a G-protein-coupled receptor with its own distribution and physiology. The H1 receptor is the one most familiar to the general public — it is the receptor blocked by Benadryl, Claritin, Allegra, Zyrtec, and the entire allergy-relief drug class.
H1 receptors are expressed on vascular endothelium, airway smooth muscle, sensory neurons, and the central nervous system. H1 activation produces:
- Vasodilation and increased vascular permeability — the wheal and flare reaction of urticaria, the runny nose of hay fever, and the systemic hypotension of anaphylaxis
- Bronchoconstriction — a contributor to allergic asthma alongside leukotriene-mediated mechanisms
- Itch — H1 receptors on cutaneous C-fiber sensory neurons signal the itch sensation centrally; this is why H1 blockers reduce pruritus in urticaria, atopic dermatitis, and insect bites
- Smooth muscle contraction in the gut — cramping and diarrhea component of severe food allergy
- CNS arousal and wakefulness — H1 receptors in the cortex and other wake-promoting regions support alertness
The two generations of H1 blockers differ pharmacologically primarily in their ability to cross the blood-brain barrier:
First-generation H1 blockers (diphenhydramine/Benadryl, chlorpheniramine, hydroxyzine, doxepin) are small, lipophilic molecules that readily cross into the CNS. They produce significant sedation, anticholinergic effects (dry mouth, urinary retention, constipation, blurred vision), and at high doses can cause delirium. Despite the side effects, they remain useful for acute allergic reactions and for off-label use as sleep aids (the "PM" in Tylenol PM is diphenhydramine).
Second-generation H1 blockers (loratadine/Claritin, fexofenadine/Allegra, cetirizine/Zyrtec, desloratadine/Clarinex, levocetirizine/Xyzal) are designed as poor blood-brain barrier penetrators — they are larger, more polar, and active substrates for the P-glycoprotein efflux pump at the BBB. They block peripheral H1 receptors to control allergy symptoms without producing the sedation of the first-generation drugs. Cetirizine is the modestly sedating exception in this class; fexofenadine is essentially non-sedating.
The H2 Receptor (Gastric Acid Secretion and Ulcer Drugs)
The H2 receptor is the histamine subtype most concentrated on parietal cells in the gastric mucosa, where it stimulates hydrochloric acid secretion. ECL cells in the stomach produce histamine in response to gastrin signaling from G-cells, the histamine acts in a local paracrine manner on adjacent parietal cells via H2 receptors, and the parietal cells then pump hydrogen ions into the gastric lumen via the H+/K+ ATPase — the proton pump.
This H2-mediated histamine pathway was the basis for one of the most consequential drug discoveries of the 20th century. Sir James Black, who had previously developed the first beta-blocker (propranolol) for cardiovascular disease, applied the same receptor-blockade approach to the H2 histamine receptor. His team at Smith Kline & French developed cimetidine (Tagamet), launched in 1976 as the first H2 blocker for peptic ulcer disease. Within a few years, cimetidine displaced surgery as the dominant treatment for peptic ulcer disease and became one of the first billion-dollar drugs in the pharmaceutical industry. Black received the Nobel Prize in Physiology or Medicine in 1988 for the development of both propranolol and cimetidine — the only Nobel ever awarded primarily for drug discovery in receptor pharmacology.
The H2 blocker class expanded with ranitidine (Zantac), famotidine (Pepcid), and nizatidine (Axid), all of which improved on cimetidine's side effect profile (cimetidine inhibits CYP450 enzymes and causes drug interactions; later H2 blockers do not). Ranitidine was the most prescribed drug in the world in 1988 before being supplanted by the proton pump inhibitor class (omeprazole/Prilosec, esomeprazole/Nexium) which block acid secretion at the final common pathway (the H+/K+ ATPase) downstream of H2.
Ranitidine was withdrawn from the US market in 2020 because of contamination with NDMA, a probable human carcinogen formed by drug degradation. Famotidine has become the dominant H2 blocker for current OTC and prescription use.
Beyond acid secretion, H2 receptors are also expressed on T cells (where they modulate Th1/Th2 balance), cardiac muscle (where they contribute to positive inotropic and chronotropic effects of histamine), and uterine smooth muscle. This is why H2 blockers occasionally have utility as adjuncts in chronic urticaria management, often combined with H1 blockers.
The H3 Autoreceptor (CNS Arousal, Narcolepsy, ADHD)
The H3 receptor was discovered later than H1 and H2 (cloned in 1999) and operates primarily as a presynaptic autoreceptor on histaminergic neurons in the central nervous system. When histamine is released into the synaptic cleft, it activates H3 receptors on the same presynaptic terminal, providing negative feedback that limits further histamine release. H3 receptors also function as heteroreceptors on serotonergic, dopaminergic, and noradrenergic neurons, where they similarly inhibit neurotransmitter release.
The pharmacological implication is that H3 inverse agonists (drugs that block the constitutive H3 activity) increase histamine, dopamine, norepinephrine, and serotonin release in the brain, producing a wake-promoting and cognition-enhancing effect. This pharmacology has been targeted for narcolepsy and excessive daytime sleepiness.
Pitolisant (Wakix) is the first marketed H3 inverse agonist, approved by the FDA in 2019 for excessive daytime sleepiness in narcolepsy. Unlike modafinil and the amphetamine-based wakefulness drugs, pitolisant is non-stimulant, non-scheduled, and has no documented abuse liability. It is now first-line therapy for narcolepsy in many European countries.
H3 inverse agonists have also been explored in clinical trials for ADHD, Alzheimer's disease cognitive symptoms, schizophrenia negative symptoms, and obesity. The development pipeline has been complicated — several candidates failed in late-stage trials — but the receptor remains an active CNS drug target.
The H4 Receptor (Pruritus, Chronic Inflammation, IBD)
The H4 receptor was the last of the four histamine receptors to be identified (cloned in 2000) and remains the most pharmacologically immature. H4 is expressed predominantly on cells of hematopoietic origin — eosinophils, mast cells, dendritic cells, T cells, and basophils — and signals chemotactic recruitment and activation of these cells to sites of inflammation.
H4 is now recognized as a key mediator of chronic pruritus that is refractory to H1 blockade. In atopic dermatitis, for example, H1 blockers like cetirizine often fail to control itch despite suppressing the wheal-and-flare response — the residual itch is mediated through H4 signaling on sensory neurons and on dermal immune cells. Selective H4 antagonists are in clinical development for atopic dermatitis, chronic spontaneous urticaria, asthma, and inflammatory bowel disease.
Adriforant (ZPL-3893787) and toreforant are two H4 antagonists that have advanced through phase 2 trials for atopic dermatitis with modest but meaningful efficacy signals. The class has not yet produced a marketed drug, but the receptor remains an active target for several pharmaceutical companies as of the mid-2020s.
DAO and HNMT: The Two Histamine Degradation Enzymes
For most signaling molecules, rapid degradation is as important as rapid release — otherwise the signal persists too long and produces pathological effects. Histamine is degraded by two enzymes with distinct tissue distributions and substrate access:
Diamine oxidase (DAO) is the dominant histamine-degrading enzyme in the intestinal lumen and at the apical surface of enterocytes. It also circulates in plasma, where it degrades histamine that has been absorbed from the gut or released into circulation during systemic anaphylaxis. DAO is a copper-containing enzyme that uses oxygen as electron acceptor and produces hydrogen peroxide and an aldehyde as products. It requires vitamin B6 and copper as essential cofactors.
DAO activity in the gut is the body's primary defense against dietary histamine. Foods naturally high in histamine (aged cheeses, cured meats, fermented vegetables, alcohol, certain fish) deliver large boluses of histamine to the intestinal lumen, and adequate DAO activity at the brush border degrades most of this before it can be absorbed. Pharmaceutical DAO inhibitors (clavulanic acid in Augmentin, isoniazid, certain MAO inhibitors) can therefore precipitate symptoms of histamine intolerance in susceptible individuals.
Genetic variation in the AOC1 gene (which codes for DAO) produces inter-individual differences in baseline DAO activity. Several SNPs are associated with reduced enzyme activity and with self-reported histamine intolerance symptoms. Inflammatory bowel disease, celiac disease, and other intestinal inflammatory states reduce DAO production by damaging the enterocytes that produce it — one of several mechanisms by which gut inflammation increases histamine sensitivity.
Histamine N-methyltransferase (HNMT) is the dominant intracellular histamine-degrading enzyme, particularly in the central nervous system, kidney, and liver. It transfers a methyl group from S-adenosylmethionine (SAMe) to histamine, producing N-methylhistamine, which is then further oxidized by monoamine oxidase B (MAO-B) for excretion.
HNMT activity is the primary defense against CNS histamine overload. HNMT polymorphisms (notably the Thr105Ile variant) reduce enzyme activity and have been associated with attention-deficit/hyperactivity disorder, multiple sclerosis, and Parkinson's disease in some studies. The role is more inferential than mechanistically proven, but the receptor pharmacology is consistent — reduced HNMT means increased CNS histamine, which means increased H3 autoreceptor activation, which means altered dopaminergic and serotonergic signaling.
The DAO/HNMT division of labor is functionally important: dietary histamine is handled by DAO at the gut barrier; endogenous histamine released into tissues is handled by HNMT inside cells. Individuals with reduced activity of either enzyme have a corresponding pattern of histamine intolerance — gut-driven symptoms in DAO deficiency, neurological and chronic inflammatory symptoms in HNMT deficiency.
Mast Cell Activation Syndrome (MCAS)
Mast cell activation syndrome (MCAS) is a clinical condition first formally described in 2007 in which patients experience episodic or chronic symptoms of mast cell mediator release without meeting criteria for systemic mastocytosis (the bone marrow disorder of clonal mast cell proliferation). MCAS is increasingly recognized as a contributor to chronic multisystem symptoms in a substantial fraction of patients with dysautonomia, Ehlers-Danlos syndrome, postural orthostatic tachycardia syndrome (POTS), and irritable bowel syndrome.
The diagnostic criteria require:
- Episodic symptoms consistent with mast cell mediator release in at least two organ systems (skin, GI, cardiovascular, respiratory, neurological)
- Biochemical evidence of mast cell activation (elevated serum tryptase during a flare, elevated urinary histamine metabolites, elevated urinary prostaglandin D2 metabolites, or elevated urinary leukotriene E4)
- Response to mast-cell-targeted therapy (H1 blockers, H2 blockers, mast cell stabilizers, leukotriene inhibitors)
The treatment approach is empiric and stepwise:
- First-line: non-sedating H1 blocker (cetirizine 10 mg or fexofenadine 180 mg daily, doubled or tripled if needed)
- Add H2 blocker: famotidine 20-40 mg twice daily for GI symptoms and combined cutaneous control
- Add mast cell stabilizer: oral cromolyn 200 mg four times daily (poorly absorbed, works at gut mucosa) or quercetin 500-1000 mg twice daily for systemic stabilization
- Add leukotriene antagonist: montelukast 10 mg daily for asthmatic component or chronic urticaria
- Refractory cases: omalizumab (anti-IgE biologic), ketotifen, vitamin C 1-2g daily as cofactor for DAO synthesis
From the histidine angle, patients with MCAS often benefit from a low-histamine diet (see below) and may benefit from DAO enzyme supplements taken with meals. Conversely, MCAS patients should generally avoid histidine supplementation, which provides additional substrate for the histamine production their mast cells are already producing in excess.
The Low-Histamine Diet for Chronic Urticaria and Intolerance
A low-histamine diet is a therapeutic eating pattern designed to reduce the dietary histamine load and the load of histamine-releasing (histamine-liberator) foods. It is used as adjunct therapy for chronic spontaneous urticaria, MCAS, and clinically diagnosed histamine intolerance.
Foods naturally high in pre-formed histamine (largely due to bacterial decarboxylation of histidine during fermentation, aging, or improper storage):
- Aged cheeses (parmesan, blue cheese, gouda, gruyere)
- Cured meats (salami, prosciutto, pepperoni, sausages)
- Fermented foods (sauerkraut, kimchi, kombucha, yogurt, kefir, miso, soy sauce)
- Alcoholic beverages, especially wine and beer
- Fish that was not flash-frozen immediately on catch — tuna, mackerel, mahi-mahi, sardines, anchovies (scombroid poisoning is essentially histamine poisoning from spoiled fish)
- Vinegar and vinegar-containing foods (pickles, ketchup, mustard)
- Avocado, spinach, eggplant, tomatoes (moderate)
Histamine-liberator foods that don't contain histamine themselves but trigger mast cell release:
- Citrus fruits, strawberries, pineapple, kiwi, banana, papaya
- Chocolate and cocoa
- Tomatoes, spinach (also high in pre-formed histamine)
- Shellfish
- Egg whites (uncooked or partially cooked)
- Nuts (especially walnuts and cashews)
- Food additives: tartrazine, benzoates, sulfites, MSG
The diet is typically implemented as a 2-4 week strict elimination followed by structured reintroduction. Symptom diaries during reintroduction help identify the specific high-histamine and liberator foods that drive symptoms for the individual patient.
Importantly, the low-histamine diet is intentionally not a long-term dietary pattern — it is excessively restrictive, eliminates many otherwise-healthy fermented foods, and risks nutritional inadequacy if continued indefinitely. The goal is to identify problem foods, treat the underlying histamine dysregulation, and gradually liberalize the diet as DAO activity improves and mast cells stabilize.
A Brief History of the Antihistamine Drug Class
Histamine was first isolated from ergot fungi in 1907 by Henry Dale and his team at the Wellcome Physiological Research Laboratories. Dale would go on to characterize histamine's role in anaphylaxis, vasodilation, and gastric acid secretion across decades of work, eventually receiving the Nobel Prize in 1936 (shared with Otto Loewi for separate work on chemical neurotransmission). Dale's recognition that an endogenous compound could mediate anaphylaxis was foundational — before his work, allergy was understood as something inherent to the "sensitized" tissue with no specific chemical mediator.
The first H1 antihistamine, piperoxan, was developed in 1937 by Daniel Bovet at the Pasteur Institute in Paris. Bovet would receive the 1957 Nobel Prize in Physiology or Medicine partly for this work. Diphenhydramine (Benadryl) was developed by George Rieveschl at the University of Cincinnati in 1943 and became one of the first widely-prescribed antihistamines, beginning a treatment era for allergic conditions that had previously been managed only with epinephrine and avoidance.
The discovery that diphenhydramine and the first-generation antihistamines crossed the blood-brain barrier and caused sedation led to two parallel developments: the use of these drugs as off-label sleep aids (still the active ingredient in most OTC sleep aids like ZzzQuil and Tylenol PM), and the search for non-sedating second-generation alternatives. Terfenadine (Seldane) was the first non-sedating H1 blocker, launched in 1985, but was withdrawn in the late 1990s due to cardiac arrhythmia risk from CYP3A4 inhibition. Its active metabolite fexofenadine (Allegra) was developed as a safer replacement and is now one of the dominant second-generation antihistamines worldwide.
The parallel discovery of H2 receptors by Sir James Black's team and the launch of cimetidine in 1976 created the H2 blocker drug class. The subsequent discovery of H3 and H4 receptors in the late 1990s and early 2000s opened the next generation of receptor-targeted histamine drugs, leading to the marketed pitolisant for narcolepsy and ongoing clinical development of H4 antagonists for chronic inflammatory conditions.
Across nearly a century of work, the histamine signaling system has produced the most pharmacologically tractable and commercially successful drug class in receptor pharmacology — a remarkable trajectory that traces back to a single amino acid (histidine) and a single enzyme (HDC).
Key Research Papers
- Dale HH, Laidlaw PP (1910). The physiological action of beta-iminazolylethylamine. Journal of Physiology. The original histamine characterization paper. — PubMed
- Bovet D, Staub AM (1937). Action protectrice des ethers phenoliques au cours de l'intoxication histaminique. Comptes Rendus de la Societe de Biologie. First antihistamine. — PubMed
- Black JW et al. (1972). Definition and antagonism of histamine H2-receptors. Nature. — PubMed
- Arrang JM et al. (1983). Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. — PubMed
- Oda T et al. (2000). Molecular cloning and characterization of a novel type of histamine receptor preferentially expressed in leukocytes. Journal of Biological Chemistry. H4 receptor identification. — PubMed
- Maintz L, Novak N (2007). Histamine and histamine intolerance. American Journal of Clinical Nutrition. The reference review on DAO/HNMT and histamine intolerance. — PubMed
- Akin C et al. (2010). Mast cell activation syndrome: Proposed diagnostic criteria. Journal of Allergy and Clinical Immunology. — PubMed
- Schwartz JC (2011). The histamine H3 receptor: from discovery to clinical trials with pitolisant. British Journal of Pharmacology. — PubMed
- Thurmond RL et al. (2008). The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nature Reviews Drug Discovery. — PubMed
- McNeil BD et al. (2015). Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature. MRGPRX2 discovery. — PubMed
- Comas-Baste O et al. (2020). Histamine intolerance: the current state of the art. Biomolecules. — PubMed
- Schnedl WJ, Enko D (2021). Histamine intolerance originates in the gut. Nutrients. — PubMed
PubMed Topic Searches
- PubMed: Histidine decarboxylase and mast cells
- PubMed: MCAS diagnosis and treatment
- PubMed: DAO and histamine intolerance
- PubMed: HNMT genetics
- PubMed: Low-histamine diet for urticaria
Connections
- Histidine Overview
- Histidine Benefits Hub
- Histidine for Hemoglobin
- Histidine and Carnosine Antioxidant Defense
- Histidine for Wound and Joint Health
- All Amino Acids
- Vitamin B6 (HDC Cofactor)
- Copper (DAO Cofactor)
- Vitamin C (Mast Cell Stabilizer)
- Eczema (Atopic Dermatitis)
- Asthma
- POTS
- IBS
- Quercetin (Mast Cell Stabilizer)
- Stinging Nettle