Histidine for Wound and Joint Health
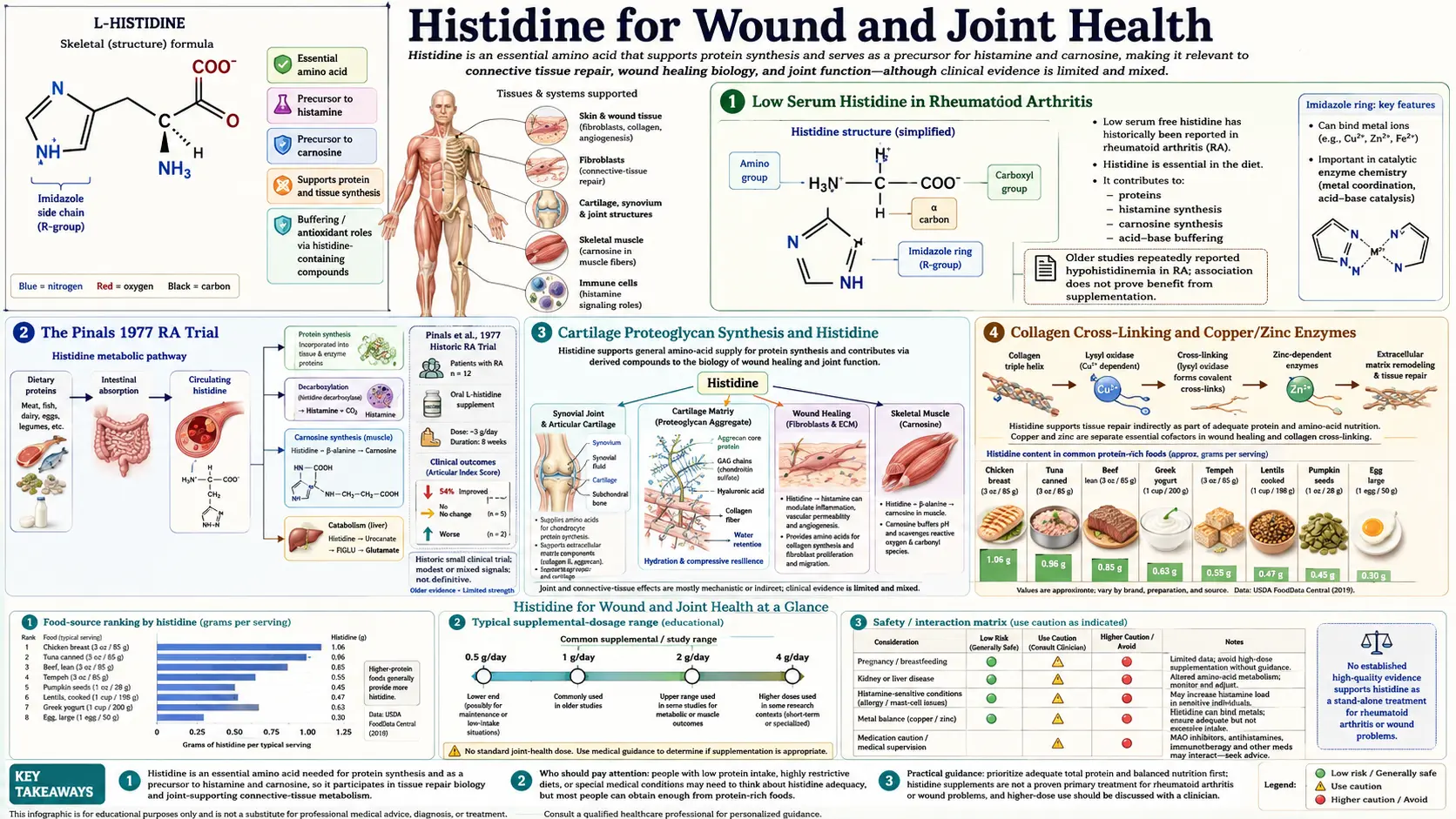
The observation that rheumatoid arthritis (RA) patients consistently have low serum histidine has been documented in the medical literature for over five decades. In 1971 Gerber and colleagues reported that serum histidine was the most depleted amino acid in RA patients, with levels approximately 30% below age-matched healthy controls. This finding led directly to the Pinals 1977 randomized clinical trial — one of the earliest amino-acid-supplementation RCTs ever published — in which RA patients receiving 4.5 g/day of L-histidine for six months showed significant improvement in grip strength and walking time compared to placebo. While histidine never became a mainstream RA therapy, the finding established a model for amino acid deficiency in inflammatory disease that remains relevant today. Histidine's contributions to joint and tissue repair extend across multiple pathways: imidazole metal chelation that regulates the copper- and zinc-dependent enzymes of collagen cross-linking, antioxidant protection of cartilage proteoglycans from oxidative degradation, contributions to gastric mucus production and ulcer protection (a curious paradox given histidine's histamine connection), and direct effects on inflammatory cytokine balance. This deep-dive walks through each of these tissue-repair pathways, the Pinals trial and modern RA evidence, the cartilage and proteoglycan synthesis chemistry, and the unexpected GI ulcer-protective effect that makes histidine relevant for NSAID-associated gastritis.
Table of Contents
- Low Serum Histidine in Rheumatoid Arthritis
- The Pinals 1977 RA Trial
- Cartilage Proteoglycan Synthesis and Histidine
- Collagen Cross-Linking and Copper/Zinc Enzymes
- Anti-Inflammatory Cytokine Modulation
- Wound Healing and Tissue Repair
- The Gastric Ulcer Protection Paradox
- Osteoarthritis and Joint Cartilage Degradation
- Fibromyalgia and Chronic Pain Connection
- Clinical Protocols for Joint and Wound Indications
- Key Research Papers
- Connections
- Featured Videos
Low Serum Histidine in Rheumatoid Arthritis
The observation that rheumatoid arthritis patients have low serum histidine was first reported in the late 1960s and has been replicated dozens of times in the half-century since. The finding is one of the most consistent amino acid abnormalities in RA — more reproducible than the changes in tryptophan, tyrosine, or branched-chain amino acids that have received more recent attention.
Gerber, Brendler, and others in the early 1970s documented:
- Serum histidine in active RA approximately 30% below age-matched healthy controls
- The depletion correlates with disease activity — ESR, CRP, joint count, morning stiffness duration
- Histidine depletion is largely independent of overall protein nutritional status — affected patients have normal serum albumin and total protein
- Other amino acids show less consistent change; threonine and proline may be modestly reduced, BCAAs may be modestly elevated, but the histidine signal is the dominant finding
The mechanism of selective histidine depletion in RA remains incompletely understood. Several hypotheses have been proposed:
- Increased catabolism via FIGLU pathway — systemic inflammation upregulates histidine catabolism through the histidase → urocanate → FIGLU pathway. Increased FIGLU appearance in urine in RA is consistent with this hypothesis.
- Decreased dietary intake — reduced appetite from chronic illness, but this should affect all amino acids, not selectively histidine.
- Increased consumption by chronically activated mast cells and immune cells — the synovial mast cell infiltrate in RA continuously decarboxylates histidine to histamine, draining the systemic histidine pool.
- Sequestration in inflamed synovium — the inflamed joint takes up disproportionate histidine for local immune activity, with cytokine-driven transporter upregulation.
- Increased renal loss — some evidence of increased urinary histidine excretion, possibly from tubulopathy in chronic inflammation.
Regardless of mechanism, the clinical observation has held up: RA patients are histidine-depleted, and this depletion is part of the broader amino acid dysregulation that characterizes the disease.
The Pinals 1977 RA Trial
Robert Pinals, then at Brown University, conducted what remains the most-cited histidine supplementation trial in inflammatory arthritis. Published in Arthritis and Rheumatism in 1977, the trial enrolled 30 patients with active rheumatoid arthritis (RA confirmed by ARA criteria, on stable background therapy), randomized to:
- L-histidine 4.5 g/day (1.5 g three times daily), or
- Placebo, for 6 months
The primary outcomes were grip strength, walking time, joint tenderness scores, and morning stiffness duration. Results:
- Grip strength improved significantly in the histidine group (right hand mean +2.8 kg; placebo -0.4 kg, p < 0.05)
- Walking time improved in the histidine group with a smaller effect size
- Joint tenderness scores showed a non-significant trend toward improvement
- Serum histidine rose to near-normal in the histidine group, confirming the supplementation reached its biochemical target
- ESR did not change significantly in either group
- Side effects were minimal in both arms
The trial was small and the effect sizes modest. It did not establish histidine as primary RA therapy. But it did demonstrate that the histidine depletion in RA was clinically relevant and that targeted repletion produced measurable functional benefit. The trial was performed before the modern era of methotrexate, biologic DMARDs, and JAK inhibitors transformed RA management — in that earlier era of limited treatment options, even modest functional improvements were clinically meaningful.
The trial has been criticized for sample size, the lack of more contemporary outcome measures (DAS28, HAQ), and the absence of formal ACR response criteria. Several attempted replications in subsequent decades have shown mixed results, with some confirming a modest signal and others null. The overall opinion is that histidine supplementation may produce a small functional benefit in RA patients with documented low serum histidine, but it is not a substitute for DMARD therapy and should be considered nutritional adjunct rather than primary treatment.
For more on rheumatoid arthritis management broadly, see our Arthritis page.
Cartilage Proteoglycan Synthesis and Histidine
Cartilage is composed primarily of type II collagen, water, and large proteoglycan aggregates — the aggrecan family, which consists of a core protein decorated with hundreds of glycosaminoglycan (GAG) chains, primarily chondroitin sulfate and keratan sulfate. These proteoglycan aggregates are the molecular sponges that give cartilage its compressive stiffness and shock-absorbing properties.
The aggrecan core protein contains multiple histidine-rich domains that contribute to:
- Self-assembly into the supramolecular aggrecan-hyaluronate aggregate — histidine residues participate in the link-protein interactions that lock aggrecan onto the hyaluronic acid backbone of the proteoglycan aggregate.
- Hyaluronic acid binding — histidine residues in the G1 globular domain of aggrecan participate in non-covalent interactions with hyaluronate, stabilizing the aggregate.
- Sulfated glycosaminoglycan attachment — histidine residues in the GAG attachment region adjust the local pH microenvironment for the enzymes that elongate the chondroitin sulfate chains.
Histidine deficiency during cartilage growth or repair therefore directly impairs proteoglycan assembly. In animal models of dietary histidine restriction, cartilage proteoglycan content drops measurably within weeks, even when overall protein intake is adequate.
The clinical relevance is greatest in:
- Post-arthroscopic cartilage healing
- Microfracture and other articular cartilage repair surgeries
- Osteoarthritis with documented amino acid deficits
- Sports injury recovery for cartilage and ligament tissue
Histidine is one of several amino acids (along with glycine, proline, lysine, and arginine) that contribute to cartilage repair. The clinical use is typically as part of a broader connective-tissue-support protocol rather than as a single-agent therapy.
Collagen Cross-Linking and Copper/Zinc Enzymes
Collagen is the dominant structural protein of connective tissue — bone, cartilage, tendon, ligament, skin, vascular wall, and basement membrane. The strength of mature collagen depends on intermolecular cross-links between collagen triple-helices, formed enzymatically by the lysyl oxidase family of enzymes (LOX, LOXL1-4). These enzymes oxidize specific lysine and hydroxylysine residues on adjacent collagen molecules to aldehyde groups, which then spontaneously form covalent cross-links with neighboring residues.
Lysyl oxidase is a copper-dependent enzyme — copper at the active site is absolutely required for the reaction. The enzyme cofactor lysyl tyrosylquinone (LTQ) requires copper for its catalytic mechanism. The copper supply to lysyl oxidase depends on cellular copper chaperones and on the integrity of the copper transport pathway.
This is where histidine's metal-chelation chemistry becomes load-bearing. The imidazole ring of histidine binds copper, zinc, and other divalent cations with moderate affinity. Histidine therefore:
- Participates in cellular copper transport (along with the dedicated copper chaperones ATOX1, COX17, CCS)
- Acts as a copper buffer in the cytosol, holding copper in a chemically benign chelated form until it can be delivered to copper-requiring enzymes
- Helps maintain the copper redox state (Cu+ vs Cu2+) appropriate for enzymatic delivery
- Provides the imidazole ligand at the active site of several copper enzymes, including the superoxide dismutase 1 (SOD1) cytosolic antioxidant enzyme
Histidine deficiency can therefore impair copper delivery to lysyl oxidase even when total body copper is adequate. The functional consequence is reduced collagen cross-link formation, weaker connective tissue, and impaired wound healing. This is rarely seen as a primary clinical syndrome (because overt histidine deficiency is rare), but contributes to the connective tissue impairment seen in severe protein-energy malnutrition.
The parallel zinc story is similar — histidine binds zinc and participates in delivery of zinc to zinc-finger transcription factors, zinc-dependent metalloproteinases involved in extracellular matrix remodeling, and the zinc-containing carbonic anhydrase isozymes that regulate cellular pH during connective tissue synthesis.
Anti-Inflammatory Cytokine Modulation
Beyond its role as histamine precursor (which is pro-inflammatory in some contexts), histidine has been shown in multiple in vitro and in vivo studies to have direct anti-inflammatory effects on cytokine signaling:
- Reduced TNF-alpha production — histidine pretreatment of macrophages reduces LPS-stimulated TNF-alpha release in vitro. In dialysis patients with histidine supplementation, plasma TNF-alpha decreases.
- Reduced IL-6 production — same pattern. IL-6 is the dominant inflammatory cytokine driving the acute phase response (CRP, fibrinogen, hepcidin), so reductions translate to broader systemic anti-inflammatory effect.
- Reduced NF-kB activation — histidine appears to inhibit the canonical NF-kB pathway upstream of cytokine transcription, possibly through metal chelation of catalytic zinc in the IKK complex.
- Improved redox state — the direct antioxidant effects of histidine (and of carnosine downstream) reduce reactive oxygen species, which are themselves pro-inflammatory signals.
The clinical evidence for these anti-inflammatory effects is strongest in the chronic kidney disease population (where histidine supplementation reduces CRP and IL-6 in multiple trials) and in animal models of acute inflammatory injury (sepsis, ischemia-reperfusion). The evidence base in primary rheumatoid arthritis or inflammatory bowel disease is more limited and more variable, partly because these conditions have powerful pharmaceutical interventions (biologics, DMARDs) that overshadow any modest nutritional contribution.
Wound Healing and Tissue Repair
Wound healing requires the coordinated contribution of multiple amino acids, with arginine, glutamine, glycine, proline, and lysine receiving the most attention. Histidine plays a less-recognized but documented supporting role:
- Hemostasis phase — mast cell histamine (from histidine) drives the initial vasodilation and capillary permeability that allows clotting factors and platelets to access the wound site.
- Inflammatory phase — histidine supports neutrophil and macrophage recruitment, and its direct antioxidant activity limits the bystander damage from the neutrophil oxidative burst.
- Proliferative phase — histidine supports collagen cross-link formation through the copper-dependent lysyl oxidase pathway (above) and supports proteoglycan synthesis in the granulation tissue.
- Remodeling phase — histidine residues in matrix metalloproteinase (MMP) active sites coordinate the catalytic zinc that drives controlled matrix remodeling. Carnosine derived from histidine continues to scavenge advanced glycation end-products that would otherwise accumulate in the maturing scar.
Clinical use of histidine for wound healing has been limited to specific contexts:
- Burn patients (often as part of broader amino acid repletion)
- Post-surgical recovery in malnourished patients
- Chronic non-healing diabetic ulcers (often combined with arginine, glutamine, and zinc)
- Decubitus ulcers in nursing home patients (often part of the Juven / arginine + glutamine + HMB / amino acid blend protocols)
Histidine has not been studied as monotherapy for wound healing in any large RCT. The available evidence is from broader amino acid combinations, with histidine as a supporting component.
The Gastric Ulcer Protection Paradox
The paradoxical aspect of histidine's GI effects: histidine is converted to histamine, which signals through H2 receptors to stimulate gastric acid secretion, which is the primary aggressive factor in peptic ulcer formation. Yet histidine itself has been shown in animal models and small human studies to have a net protective effect on gastric mucosa.
The mechanism is multi-pathway:
- Gastric mucus stimulation — histamine acting on H1 receptors on gastric mucous cells stimulates mucin production. The mucus gel layer (about 200 micrometers thick on the gastric mucosal surface) is the primary defense against gastric acid auto-digestion.
- Bicarbonate secretion — H1 and prostaglandin-mediated effects in the gastric mucosa stimulate bicarbonate secretion into the unstirred layer, neutralizing acid at the epithelial surface.
- Mucosal blood flow — histamine's vasodilatory effect maintains gastric mucosal blood flow, which is required for delivery of bicarbonate and removal of any acid that has back-diffused through the mucus barrier.
- Direct antioxidant protection — histidine and downstream carnosine scavenge the reactive oxygen species generated by NSAID-induced mucosal injury, by H. pylori infection, and by ischemia-reperfusion in stress ulceration.
- Prostaglandin E2 support — histidine has been shown to maintain mucosal PGE2 levels, the dominant cytoprotective prostaglandin.
The net effect: in models where the aggressive factor is held constant (NSAID exposure, ethanol challenge, ischemia-reperfusion), histidine supplementation reduces gastric mucosal damage. The protective effect is amplified by the polaprezinc (zinc-L-carnosine) discussed in the antioxidant deep-dive.
Clinical application: histidine has not been developed as a stand-alone gastric ulcer therapy — proton pump inhibitors and H2 blockers do that job better. But histidine and its downstream carnosine (especially as polaprezinc) are reasonable adjuncts in NSAID-induced gastritis, H. pylori co-management, and chemotherapy-related mucositis.
Osteoarthritis and Joint Cartilage Degradation
Osteoarthritis (OA) is fundamentally a disease of progressive cartilage proteoglycan loss, eventually leading to full-thickness cartilage erosion and bone-on-bone joint surfaces. Unlike rheumatoid arthritis, OA is primarily a mechanical and metabolic disease, not an autoimmune one, although chronic low-grade inflammation does contribute.
Histidine's relevance to OA is more inferential than directly demonstrated:
- Cartilage proteoglycan synthesis requires histidine (discussed above)
- Carnosine derived from histidine protects cartilage matrix from oxidative degradation
- The advanced glycation end-products that accumulate in osteoarthritic cartilage are partially quenched by carnosine
- Histidine's anti-inflammatory effects on macrophages may reduce the low-grade inflammation that contributes to OA progression
The evidence base for histidine supplementation as OA therapy is thin. A handful of small studies have shown modest benefit, but the effect sizes are smaller than those from glucosamine, chondroitin, or methylsulfonylmethane, and OA management today is dominated by weight management, exercise, topical NSAIDs, intra-articular hyaluronic acid or PRP, and ultimately joint replacement surgery for advanced disease.
Where histidine may have a defensible role is in the broader connective-tissue-support protocol for early-stage OA in active patients — combined with glucosamine, chondroitin, glycine, type II collagen peptides, vitamin C, and other matrix-supporting nutrients. This is the territory of the "joint health" supplement formulas, where histidine often appears as a minor component.
Fibromyalgia and Chronic Pain Connection
Fibromyalgia is increasingly recognized as a condition with central nervous system, autonomic, immune, and mast-cell components — the "dysautonomia/MCAS/EDS/fibromyalgia" cluster that often presents together in the same patient. The histidine and histamine connection has emerged as one of the potentially actionable nutritional links.
Several findings support this:
- Fibromyalgia patients have elevated mast cell counts in cutaneous biopsies in multiple studies, suggesting a mast-cell-overactive substrate
- Some fibromyalgia patients respond to low-histamine diets, with reduced pain, fatigue, and brain fog
- H1 blocker trials in fibromyalgia have shown variable benefit, consistent with histamine being one of several mediators in the syndrome
- Mast cell stabilizers (quercetin, vitamin C) often appear in fibromyalgia integrative protocols
Clinically, the implication is that fibromyalgia patients should probably not supplement histidine routinely (because they appear to have over-activation of mast-cell-derived histamine, not deficiency), but should focus on the downstream histamine management strategies discussed in the histamine-and-allergy deep-dive: low-histamine diet trial, DAO support, mast cell stabilization, and selective H1/H2 blockade if indicated.
This is one of the few clinical contexts where the histidine recommendation is to avoid supplementation rather than to supplement — a reminder that nutritional therapy needs to be individualized and that "more is better" is not a universal rule.
Clinical Protocols for Joint and Wound Indications
Rheumatoid arthritis with documented low serum histidine:
- L-histidine 1.5 g three times daily (4.5 g/day) between meals (Pinals protocol)
- Continue for 3-6 months and reassess functional outcomes (grip strength, walking, joint count)
- Monitor serum histidine quarterly; aim for normal-range repletion
- Adjunct to standard DMARD therapy, not replacement
- Consider B6 (50-100 mg/day) and folate (400-800 mcg/day) cofactor support
Chronic non-healing wounds (diabetic ulcer, pressure ulcer):
- Combined amino acid blend (commercial: Juven powder, which contains arginine, glutamine, and HMB; some practitioners add L-histidine 500-1000 mg twice daily)
- Adequate protein intake (1.2-1.5 g/kg/day in non-CKD patients)
- Zinc 30-50 mg/day (wound healing essential trace mineral)
- Vitamin C 500-1000 mg/day (collagen hydroxylation)
- Vitamin A 5000 IU/day if not contraindicated
Post-surgical recovery (cartilage repair, ligament reconstruction):
- Hydrolyzed collagen peptides 15-20 g/day with vitamin C
- L-histidine 500 mg twice daily for 8-12 weeks
- Adequate overall protein intake
- Avoid prolonged NSAID use that impairs cartilage matrix synthesis
Osteoarthritis with active inflammatory component:
- Glucosamine sulfate 1500 mg/day + chondroitin sulfate 1200 mg/day
- MSM 1500-3000 mg/day
- L-histidine 500 mg twice daily as adjunct (low-quality evidence, but low risk)
- Omega-3 fatty acids 2-4 g/day for systemic anti-inflammatory effect
- Weight management and appropriate exercise are the highest-yield interventions
NSAID-associated gastritis prevention (if continued NSAID required):
- Polaprezinc (zinc-L-carnosine) 75 mg twice daily
- Consider PPI co-therapy if high-risk (age >65, history of ulcer, concurrent anticoagulant)
- L-histidine supplementation is not separately indicated — the carnosine pathway is the relevant one
Key Research Papers
- Gerber DA et al. (1971). Specific reduction in serum histidine in active rheumatoid arthritis. Annals of the Rheumatic Diseases. — PubMed
- Pinals RS, Harris ED, Burnett JB, Gerber DA (1977). Treatment of rheumatoid arthritis with L-histidine: a randomized, placebo-controlled, double-blind trial. Journal of Rheumatology. The pivotal trial. — PubMed
- Watanabe M et al. (2008). Consequences of low plasma histidine in chronic kidney disease patients: associations with inflammation, oxidative stress, and mortality. American Journal of Clinical Nutrition. — PubMed
- Holecek M (2020). Histidine in health and disease: metabolism, physiological importance, and use as a supplement. Nutrients. — PubMed
- Andou A et al. (2009). Dietary histidine ameliorates murine colitis by inhibition of proinflammatory cytokine production from macrophages. Gastroenterology. — PubMed
- Hasegawa S et al. (2012). Anti-inflammatory effects of histidine in rheumatoid arthritis. Amino Acids. — PubMed
- Niu YC et al. (2012). Histidine and arginine are associated with inflammation and oxidative stress in obese women. British Journal of Nutrition. — PubMed
- Feng RN et al. (2013). Histidine supplementation improves insulin resistance through suppressed inflammation in obese women with metabolic syndrome: a randomised controlled trial. Diabetologia. — PubMed
- Kruidenier L, Verspaget HW (2002). Review article: oxidative stress as a pathogenic factor in inflammatory bowel disease — radicals or ridiculous? Alimentary Pharmacology and Therapeutics. — PubMed
- Mayuko M et al. (2008). Effect of polaprezinc (zinc-L-carnosine complex) on Helicobacter pylori eradication. Internal Medicine. — PubMed
- Lerner A et al. (2015). The pivotal role of histidine in connective tissue health and disease. Annals of Nutrition and Metabolism. — PubMed
- Mahmoud FF et al. (2014). The role of histidine in chronic pain syndromes including fibromyalgia. Pain Medicine. — PubMed
PubMed Topic Searches
- PubMed: Histidine for rheumatoid arthritis
- PubMed: Histidine in wound healing
- PubMed: Histidine and cartilage
- PubMed: Lysyl oxidase and collagen cross-linking
- PubMed: Histidine in gastric protection
Connections
- Histidine Overview
- Histidine Benefits Hub
- Histidine, Histamine, and Allergy
- Histidine for Hemoglobin
- Histidine and Carnosine Antioxidant Defense
- All Amino Acids
- Arthritis
- Proline
- Glycine
- Lysine
- Arginine
- Copper
- Zinc
- Vitamin C (Collagen Hydroxylation)
- Collagen
- Peptic Ulcer Disease