Glutamic Acid for Neurotransmission — The Brain's Major Excitatory Signal
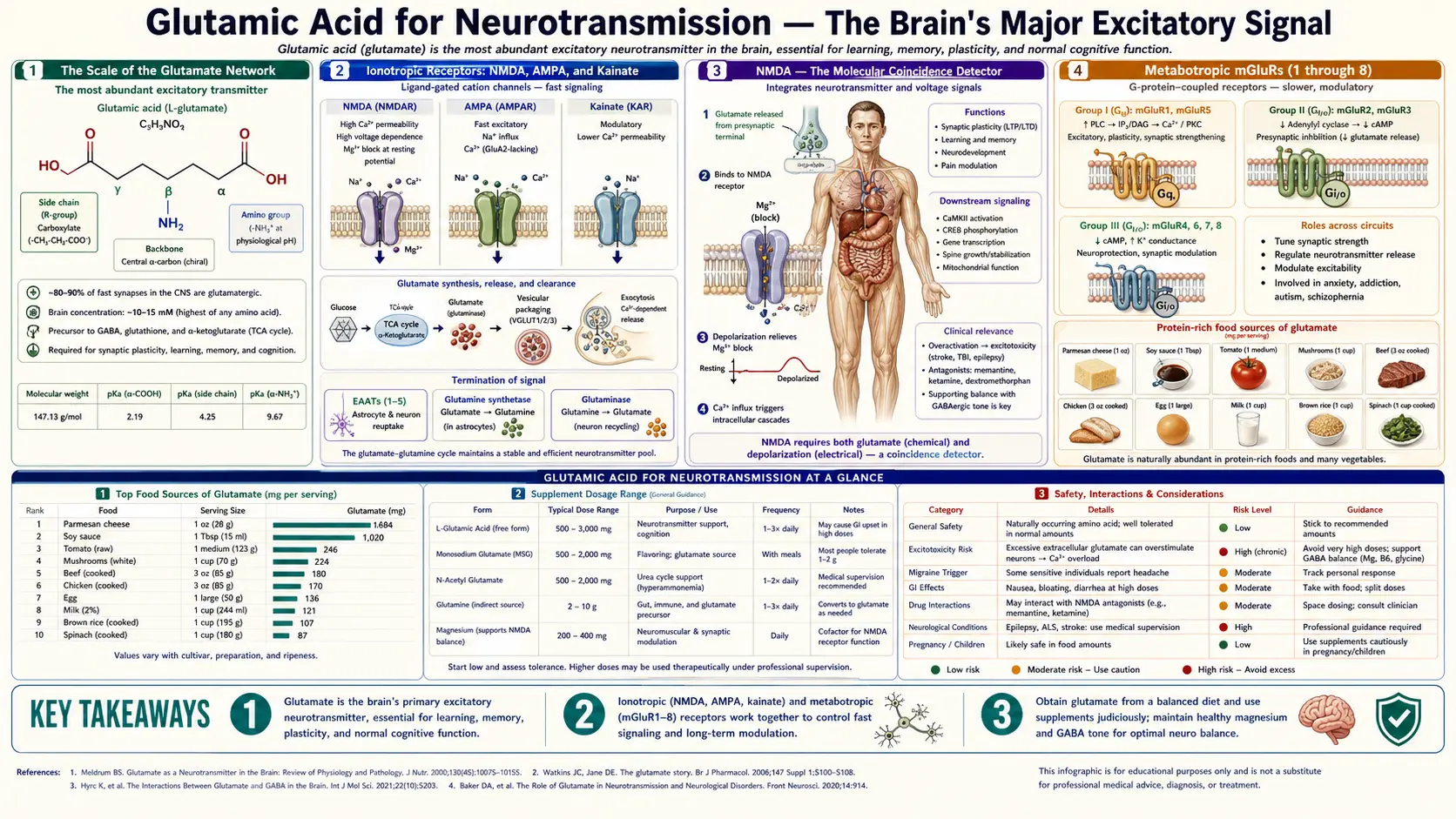
Glutamate is the principal excitatory neurotransmitter in the vertebrate central nervous system, mediating somewhere between 80% and 90% of all fast excitatory synaptic transmission and present at roughly half of all CNS synapses. Every conscious thought, voluntary movement, memory encoded into long-term storage, and sensory percept depends on glutamate binding to one of four receptor families — the ionotropic NMDA, AMPA, and kainate channels, and the metabotropic mGluR1 through mGluR8 G-protein coupled receptors. The same molecule that drives learning through NMDA-dependent long-term potentiation can, when uncleared, kill the neurons it normally activates — the excitotoxicity that underlies the secondary damage in stroke, traumatic brain injury, ALS, and Huntington's disease. The clinical drug list reads like a tour of glutamate pharmacology: memantine for Alzheimer's disease, ketamine for treatment-resistant depression and anesthesia, dextromethorphan for cough and combined with bupropion for depression, and lamotrigine for epilepsy and bipolar disorder all act on the glutamate system. This deep dive walks through the receptor biology, the synaptic mechanism of learning, the excitotoxic cascade, and the modern NMDA-antagonist drug class.
Table of Contents
- The Scale of the Glutamate Network
- Ionotropic Receptors: NMDA, AMPA, and Kainate
- NMDA — The Molecular Coincidence Detector
- Metabotropic mGluRs (1 through 8)
- Long-Term Potentiation and the Cellular Basis of Learning
- The Glutamate-Glutamine Cycle (Astrocyte Recycling)
- Excitotoxicity — When the Brain's Major Signal Kills
- Stroke and Traumatic Brain Injury
- ALS and Huntington's Disease
- NMDA Antagonist Drugs (Memantine, Ketamine, DXM)
- Magnesium as the Natural NMDA Blocker
- Key Research Papers
- Connections
- Featured Videos
The Scale of the Glutamate Network
To appreciate why glutamate matters so much, start with the numbers. The adult human brain contains approximately 86 billion neurons connected by an estimated 100 to 500 trillion synapses. Roughly half of these synapses use glutamate as their neurotransmitter. By volume of synaptic activity, glutamate dwarfs every other neurotransmitter the brain uses — dopamine, serotonin, norepinephrine, acetylcholine, and GABA all play important roles, but quantitatively the brain is overwhelmingly a glutamate machine.
The concentration of glutamate inside synaptic vesicles is extraordinary: approximately 100 millimolar, the same order of magnitude as intracellular potassium. When a vesicle fuses with the presynaptic membrane, it releases a bolus of glutamate that achieves a transient concentration of about 1 millimolar in the 20-nanometer-wide synaptic cleft — enough to saturate AMPA receptors within microseconds and trigger the postsynaptic excitatory current. Extracellular glutamate is then rapidly cleared by transporter proteins to a resting concentration of approximately 1 micromolar, a thousand-fold lower than the peak. This four-order-of-magnitude swing is what makes the synapse a fast, high-fidelity information-transmission channel.
Because glutamate is also a metabolic intermediate (it sits at the crossroads of amino-acid transamination and the citric acid cycle — see the Nitrogen Metabolism page), the brain has to maintain a strict separation between its metabolic and signaling pools. The blood-brain barrier severely restricts glutamate entry from the periphery (the brain manufactures essentially all of its own glutamate from glutamine and alpha-ketoglutarate), and astrocyte transporters rapidly sequester any glutamate that escapes from synapses. This compartmentalization is the reason dietary glutamate, including MSG, has minimal direct effect on brain glutamate signaling under normal conditions — the brain's pool is essentially sealed off from the body's. See the MSG page for the full story.
Ionotropic Receptors: NMDA, AMPA, and Kainate
Glutamate acts on two structurally distinct receptor superfamilies. The ionotropic glutamate receptors (iGluRs) are ligand-gated ion channels that open within milliseconds of glutamate binding and mediate the fast excitatory postsynaptic current. They are subdivided into three families named for their selective agonists discovered decades before the receptors were cloned:
- AMPA receptors (alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) are the workhorses of moment-to-moment fast excitatory transmission. They open within sub-millisecond timescales of glutamate binding, permit sodium and potassium flux, and depolarize the postsynaptic membrane. Most AMPA receptors are calcium-impermeable due to the presence of an edited GluA2 subunit, though calcium-permeable variants exist in some interneurons. AMPA receptor density at synapses is highly dynamic — insertion and removal of AMPA receptors is the molecular substrate of synaptic plasticity. The number of AMPA receptors at a synapse fluctuates over minutes to hours in response to neural activity, and this trafficking is the physical basis of learning at the level of individual synaptic contacts.
- NMDA receptors (N-methyl-D-aspartate) require both glutamate binding and a co-agonist (glycine or D-serine) plus simultaneous postsynaptic depolarization to remove a magnesium block from the channel pore. When all three conditions are met, the channel opens and is highly permeable to calcium in addition to sodium and potassium. NMDA-mediated calcium influx is the trigger for long-term changes in synaptic strength — both potentiation and depression. NMDA receptors are heterotetramers built from NR1, NR2 (A-D), and sometimes NR3 subunits, with regional and developmental variations in subunit composition that confer different channel kinetics and pharmacological sensitivities.
- Kainate receptors are less abundant and less well understood. They act presynaptically to modulate neurotransmitter release at many synapses and postsynaptically to generate slower excitatory currents than AMPA. Kainate receptor dysfunction has been implicated in temporal lobe epilepsy, and the toxic algal compound domoic acid (the cause of amnesic shellfish poisoning) acts at kainate receptors to produce severe excitotoxic seizures and hippocampal damage.
The differential kinetics and properties of these three receptor families allow the same glutamate molecule to encode multiple types of information — fast yes/no signals (AMPA), coincidence-detected learning signals (NMDA), and modulatory tone (kainate).
NMDA — The Molecular Coincidence Detector
The NMDA receptor is one of the most elegant signaling molecules in biology. Unlike most ligand-gated channels, which open when their ligand binds, NMDA channels demand a precise combination of three events occurring within tens of milliseconds:
- Presynaptic glutamate release — the orthosteric agonist binds the NR2 subunit binding pocket
- Co-agonist binding — glycine or D-serine binds the NR1 subunit binding pocket (D-serine, produced by astrocytes from L-serine via serine racemase, is the dominant co-agonist in the forebrain; glycine dominates in the brainstem and spinal cord)
- Postsynaptic depolarization — the resting membrane potential of approximately −70 mV must rise to roughly −30 mV or above to dislodge a magnesium ion that physically plugs the open channel pore at hyperpolarized voltages
Only when all three conditions converge does the channel actually conduct ions. This three-input AND gate is why NMDA receptors are called coincidence detectors — they fire only when presynaptic activity coincides with postsynaptic activity, which is exactly what an unsupervised learning rule needs to detect. Donald Hebb's 1949 postulate that "cells that fire together wire together" turned out to have an exact molecular embodiment in the NMDA receptor 30 years later, when Tim Bliss and Terje Lomo first described long-term potentiation in the rabbit hippocampus.
The calcium that flows in through the open NMDA channel is the trigger for a cascade of intracellular signaling events: activation of calcium/calmodulin-dependent protein kinase II (CaMKII), phosphorylation of AMPA receptor subunits, recruitment of additional AMPA receptors to the synapse, and ultimately new gene transcription via CREB-dependent transcription. The result is a synapse that responds more strongly to the same presynaptic input the next time it occurs — the cellular basis of associative memory.
Metabotropic mGluRs (1 through 8)
Parallel to the fast-acting ionotropic channels, glutamate also activates a family of metabotropic glutamate receptors (mGluRs), which are seven-transmembrane G-protein-coupled receptors. There are eight subtypes (mGluR1 through mGluR8), organized into three groups based on sequence homology, G-protein coupling, and pharmacology:
- Group I (mGluR1, mGluR5) — coupled to Gq, activates phospholipase C, generates inositol trisphosphate and diacylglycerol, mobilizes intracellular calcium, and generally increases postsynaptic excitability. mGluR5 antagonists have been explored for fragile X syndrome (where excessive mGluR5 signaling drives the cognitive and behavioral phenotype) and for L-DOPA-induced dyskinesia in Parkinson's disease.
- Group II (mGluR2, mGluR3) — coupled to Gi/Go, inhibits adenylate cyclase, reduces cAMP, and typically functions presynaptically to decrease glutamate release. mGluR2/3 agonists have been studied as candidate antipsychotics and anxiolytics, though clinical trial results have been mixed.
- Group III (mGluR4, mGluR6, mGluR7, mGluR8) — also Gi/Go coupled and largely presynaptic. mGluR6 is found exclusively in retinal ON-bipolar cells and is essential for the inverted (sign-reversing) photoreceptor-to-bipolar synapse. Group III agonists are being explored for Parkinson's, anxiety, and absence epilepsy.
Whereas iGluRs operate on the millisecond timescale and generate the fast excitatory current that conveys the actual neural signal, mGluRs operate on the hundreds-of-milliseconds to seconds timescale and modulate neuronal excitability, synaptic plasticity, and neurotransmitter release. They are the volume knob and tone control rather than the audio signal itself.
Long-Term Potentiation and the Cellular Basis of Learning
Long-term potentiation (LTP) is the long-lasting strengthening of synaptic transmission that occurs after brief high-frequency stimulation of a presynaptic pathway. First described by Bliss and Lomo in 1973 in the rabbit hippocampal perforant pathway, LTP has become the most widely studied cellular model of learning and memory. The defining features of LTP are cooperativity (a threshold number of synapses must be co-activated), associativity (weak inputs can be potentiated if paired with a strong input), and input specificity (only the activated synapses are strengthened, not neighboring synapses on the same neuron).
The molecular cascade goes roughly as follows: (1) high-frequency presynaptic stimulation releases enough glutamate to strongly activate AMPA receptors and depolarize the postsynaptic membrane, (2) the depolarization removes the magnesium block from NMDA channels at the same synapse, (3) calcium floods through the open NMDA channels, (4) calcium activates CaMKII, which autophosphorylates and becomes persistently active, (5) CaMKII phosphorylates the GluA1 subunit of AMPA receptors, increasing their single-channel conductance and promoting trafficking of additional AMPA receptors to the synapse, (6) for late-phase LTP lasting beyond an hour, CREB-mediated transcription of new proteins (including BDNF, Arc, and the kinase PKMzeta) consolidates the change into a stable structural alteration of the dendritic spine.
The corresponding process for weakening synapses is long-term depression (LTD), triggered by lower-frequency presynaptic stimulation that produces smaller and more sustained calcium influxes through NMDA channels. The calcium dynamics — brief and large for LTP, prolonged and small for LTD — route the signal through different downstream kinases and phosphatases.
The hippocampal pyramidal neuron is the canonical setting for LTP study, but every cortical area and many subcortical regions express NMDA-dependent plasticity. The cellular mechanism is conserved from worm to human. Drugs that block NMDA receptors (like ketamine at low doses, dextromethorphan, or the experimental antagonist MK-801) reliably impair learning and memory formation in animals and humans — though as discussed below, the same drugs can paradoxically produce antidepressant effects through a different mechanism.
The Glutamate-Glutamine Cycle (Astrocyte Recycling)
Because extracellular glutamate must be cleared from the synapse within milliseconds and because the brain cannot afford to lose its glutamate supply to peripheral metabolism, neurons and astrocytes operate one of the most metabolically active recycling pathways in biology — the glutamate-glutamine cycle:
- The presynaptic neuron releases glutamate into the synaptic cleft
- Glutamate binds and activates postsynaptic AMPA/NMDA/kainate receptors, then diffuses laterally
- Astrocyte processes that envelop the synapse express high-affinity glutamate transporters (EAAT1/GLAST and EAAT2/GLT-1, accounting for over 90% of synaptic glutamate clearance in the adult forebrain) that take up glutamate against a steep concentration gradient using the sodium electrochemical gradient as the driving force
- Inside the astrocyte, the enzyme glutamine synthetase adds an ammonia molecule to glutamate using one molecule of ATP, producing glutamine
- Glutamine is released from the astrocyte into the extracellular space, where neurons take it up via system A and system N amino acid transporters
- Inside the neuron, the enzyme phosphate-activated glutaminase (PAG) reverses the reaction, releasing ammonia and regenerating glutamate, which is then concentrated into synaptic vesicles by vesicular glutamate transporters (VGLUT1, VGLUT2, VGLUT3)
This cycle consumes a substantial fraction of the brain's total energy budget — the brain accounts for only 2% of body weight but uses roughly 20% of resting metabolism, and a large share of that energy goes to operating the glutamate transporters and the glutamine synthetase / glutaminase enzymes. When astrocyte glutamate transport fails (as in some forms of ALS, where the EAAT2/GLT-1 transporter is downregulated, or in acute ischemia where ATP runs out and the sodium gradient collapses), extracellular glutamate accumulates to neurotoxic levels and triggers the excitotoxic cascade discussed below.
The cycle also tightly couples glutamatergic neurotransmission to ammonia metabolism — the glutamine synthetase step in astrocytes is the brain's primary ammonia detoxification pathway. In hepatic encephalopathy (where the failing liver cannot clear systemic ammonia), the resulting hyperammonemia overwhelms astrocyte glutamine synthetase, glutamine accumulates osmotically, astrocytes swell, and intracranial pressure rises. The link between glutamate biology and ammonia handling is one of the strongest examples of how brain signaling and intermediary metabolism are inseparable. See the Nitrogen Metabolism page for more detail on this connection.
Excitotoxicity — When the Brain's Major Signal Kills
The same NMDA-receptor calcium influx that drives learning can, when excessive and prolonged, kill the neuron carrying it. The phenomenon was first formally characterized by John Olney in 1969, who showed that systemic injection of glutamate or kainic acid produced selective neuronal death in regions of the brain not protected by the blood-brain barrier (the arcuate nucleus, the area postrema, the subfornical organ). He coined the term excitotoxicity to capture the paradox that the brain's major excitatory signal could itself be a neurotoxin under the right conditions.
The cell-death cascade has been mapped in detail over the subsequent five decades. Excessive NMDA-mediated calcium influx triggers:
- Activation of calcium-dependent proteases (calpains) that cleave structural proteins including spectrin and tau
- Calcium-dependent phospholipase A2 activation, liberating arachidonic acid and generating downstream eicosanoid inflammation
- Mitochondrial calcium overload, opening of the mitochondrial permeability transition pore, collapse of the inner mitochondrial membrane potential, release of cytochrome c, and apoptosome assembly
- Nitric oxide synthase (nNOS) activation, generating nitric oxide that combines with superoxide to form peroxynitrite, a highly reactive oxidant that nitrates tyrosine residues on key proteins
- DNA fragmentation through PARP-1 hyperactivation, depleting cellular NAD+ and ATP and triggering parthanatos cell death
- Direct membrane damage, edema, and necrosis when the cascade is severe enough
The neurons most vulnerable to excitotoxicity are those with the highest density of NMDA receptors and the heaviest reliance on calcium signaling — hippocampal CA1 pyramidal cells, cerebellar Purkinje cells, striatal medium spiny neurons, and cortical layer 5 pyramidal cells. This pattern of selective vulnerability maps closely to the patterns of neuronal loss in stroke, hypoglycemia, status epilepticus, traumatic brain injury, Huntington's disease, and ALS — suggesting that excitotoxicity is a final common pathway for diverse neurological insults.
Stroke and Traumatic Brain Injury
Ischemic stroke (the most common form, accounting for approximately 87% of all strokes) produces a core of infarcted tissue surrounded by a penumbra of hypoperfused but still-viable tissue. Within minutes of arterial occlusion, the penumbral neurons run out of ATP, the sodium/potassium ATPase fails, the cells depolarize, glutamate floods out of presynaptic terminals (and is reverse-transported out of astrocytes whose sodium gradient has collapsed), and extracellular glutamate concentrations rise from the resting micromolar range to the millimolar range. The penumbral neurons that survived the initial loss of blood flow now die from excitotoxic NMDA-mediated calcium overload over the subsequent hours — this is the rationale for the narrow therapeutic window for thrombolysis (tissue plasminogen activator within 4.5 hours of symptom onset) and mechanical thrombectomy.
Decades of clinical trials have tested NMDA antagonists (selfotel, eliprodil, gavestinel, aptiganel) and AMPA antagonists (talampanel) as stroke neuroprotectants. Almost all have failed in phase III despite robust efficacy in animal models. The reasons are debated: the inability to dose patients early enough, the off-target sedation and psychotomimetic effects of NMDA blockers, the heterogeneity of human stroke, and possibly the fact that some excitotoxic calcium signaling is also necessary for ischemic preconditioning and post-stroke recovery. Memantine, the partial NMDA antagonist now approved for Alzheimer's, is gentler than the older agents and remains under investigation for various neuroprotective indications.
Traumatic brain injury follows a similar pattern. The initial mechanical insult triggers a wave of cortical spreading depression and indiscriminate glutamate release, leading to secondary excitotoxic damage that extends well beyond the primary impact site. Microdialysis studies in TBI patients confirm extracellular glutamate rises 50-fold or more in the first hours after injury. Therapeutic mild hypothermia, magnesium infusion (magnesium being the natural NMDA blocker, discussed below), and avoidance of secondary injury (hypotension, hypoxia, hyperthermia) are the current evidence-based approaches to limiting secondary excitotoxic damage in moderate-to-severe TBI.
ALS and Huntington's Disease
Amyotrophic lateral sclerosis (ALS) is the textbook chronic excitotoxic disease. Motor neuron death in ALS is associated with reduced expression of the glial glutamate transporter EAAT2 (GLT-1), leading to elevated extracellular glutamate in the motor cortex and spinal cord. Motor neurons happen to express a higher proportion of calcium-permeable AMPA receptors (those lacking the edited GluA2 subunit) than most other neurons, making them especially vulnerable to AMPA-mediated calcium overload in addition to NMDA-mediated overload. The one disease-modifying drug approved for ALS in the United States, riluzole, works at least partly by reducing presynaptic glutamate release (through use-dependent sodium channel block) and stabilizing the inactivated state of voltage-gated sodium channels. Riluzole extends survival by approximately 2 to 3 months — a modest but real effect, and the only disease-modifying option for decades until the addition of edaravone (2017) and tofersen (2023, for SOD1 mutations specifically).
Huntington's disease shows a different excitotoxic flavor. The mutant huntingtin protein produces selective degeneration of striatal medium spiny neurons, which receive massive glutamatergic input from the cortex. The classical excitotoxic model of Huntington's involves enhanced NMDA-receptor signaling at extrasynaptic NMDA sites (which signal through pro-death pathways) and impaired synaptic NMDA signaling (which is pro-survival). The kynurenine pathway is dysregulated in Huntington's as well, with shifts toward production of quinolinic acid (an endogenous NMDA agonist and excitotoxin) over kynurenic acid (an endogenous NMDA antagonist and neuroprotectant). The clinical drug tetrabenazine reduces hyperkinetic movements in Huntington's by depleting presynaptic dopamine, but no disease-modifying therapy yet exists.
Alzheimer's disease also shows excitotoxic features, particularly in the glutamatergic projections from the entorhinal cortex to the hippocampus that are among the earliest affected. This is the rationale for memantine's indication in moderate-to-severe Alzheimer's — not as a cognition booster but as a damper on tonic extrasynaptic NMDA activation that contributes to ongoing neurodegeneration.
NMDA Antagonist Drugs (Memantine, Ketamine, DXM)
The NMDA receptor pharmacology bench has produced several clinically important drugs, despite the failure of pure NMDA antagonists in stroke. The key has been finding agents that block NMDA receptors strongly enough to be therapeutically useful but weakly enough to avoid the dissociative, sedative, and amnestic side effects of pure antagonists like PCP (phencyclidine) and MK-801.
- Memantine (Namenda) is a moderate-affinity, uncompetitive, voltage-dependent NMDA antagonist that preferentially blocks pathologically open channels while sparing the brief openings associated with normal synaptic transmission. Approved for moderate-to-severe Alzheimer's disease, it produces a modest but statistically significant improvement in cognitive and functional decline. The mechanism is thought to involve reduction of tonic extrasynaptic NMDA activation by ambient glutamate, sparing the phasic synaptic activation needed for learning.
- Ketamine is a non-competitive NMDA channel blocker originally developed as a dissociative anesthetic. At anesthetic doses (1-2 mg/kg IV), it produces complete dissociation and analgesia with preserved respiration — making it the preferred field anesthetic for trauma and the standard for pediatric procedural sedation. At much lower subanesthetic doses (0.5 mg/kg IV over 40 minutes), it produces a rapid (hours-to-days) antidepressant effect in treatment-resistant depression. The FDA approved the S-enantiomer esketamine as an intranasal spray (Spravato) in 2019 for treatment-resistant depression. The antidepressant mechanism is not fully understood but appears to involve transient NMDA blockade followed by a downstream surge in glutamate release, AMPA receptor activation, BDNF release, and new dendritic spine formation — a kind of forced synaptic re-wiring.
- Dextromethorphan (DXM) is the dextrorotatory isomer of the opioid levorphanol, retained in over-the-counter cough syrup for its central antitussive effect. DXM is an NMDA antagonist at moderate concentrations, which accounts for its dissociative effects at recreational doses (the "robotripping" phenomenon). The FDA approved a dextromethorphan-bupropion combination (Auvelity) in 2022 for major depressive disorder — bupropion inhibits the cytochrome P450 enzyme that metabolizes dextromethorphan, boosting its plasma levels and enabling sustained NMDA blockade at oral doses.
- Nitrous oxide ("laughing gas") is a weak NMDA antagonist used as a dental and obstetric anesthetic. A small but well-conducted trial published in Science Translational Medicine in 2015 reported a rapid antidepressant effect from a single 1-hour inhalation of 50% nitrous oxide, paralleling the ketamine literature.
- Lamotrigine is a use-dependent sodium channel blocker that reduces glutamate release presynaptically. It is approved for partial-onset seizures, generalized tonic-clonic seizures, Lennox-Gastaut syndrome, and maintenance therapy in bipolar disorder.
- Riluzole (Rilutek), discussed above for ALS, has off-label use for treatment-resistant depression and OCD on the same glutamate-modulation rationale.
The emergence of ketamine and esketamine as rapid-acting antidepressants represents one of the most important developments in psychiatric pharmacology in decades — the first new mechanism of action since the SSRIs in the late 1980s. It has put glutamate signaling at the center of psychiatric research.
Magnesium as the Natural NMDA Blocker
The endogenous, physiologic block of the NMDA channel pore is provided by magnesium, which sits in the channel at resting membrane potentials and is expelled when the cell depolarizes. This voltage-dependent magnesium block is the molecular basis of the NMDA coincidence detector property described above. Without adequate intracellular and extracellular magnesium, the NMDA receptor loses its voltage dependence, opens too easily, and admits calcium under conditions that should not normally trigger learning — or worse, conditions that should not normally trigger calcium influx at all.
This has practical clinical implications. Magnesium deficiency is widespread (estimates suggest 50% or more of the U.S. population consumes less than the RDA), and the relationship between magnesium status and central nervous system hyperexcitability has been documented in conditions including migraine, anxiety, insomnia, and seizure threshold. Magnesium repletion is one of the few nutritional interventions with reasonable evidence for migraine prophylaxis (typical dose 400-600 mg/day of magnesium glycinate or magnesium citrate) and is part of the standard preeclampsia treatment protocol (IV magnesium sulfate to reduce seizure risk). See the Magnesium page for more on dosing and forms.
For patients with conditions involving glutamatergic hyperexcitability — migraine, anxiety, insomnia, restless legs syndrome, post-concussive syndrome — ensuring adequate magnesium status is one of the simplest and safest interventions a naturopathic practitioner can recommend. The mechanism is the same one that makes magnesium the body's built-in NMDA channel guardian.
Key Research Papers
- Bliss TV, Lomo T (1973). Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. Journal of Physiology. — PubMed
- Olney JW (1969). Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. — PubMed
- Choi DW (1988). Glutamate neurotoxicity and diseases of the nervous system. Neuron. — PubMed
- Berman RM et al. (2000). Antidepressant effects of ketamine in depressed patients. Biological Psychiatry. — PubMed
- Zarate CA et al. (2006). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Archives of General Psychiatry. — PubMed
- Reisberg B et al. (2003). Memantine in moderate-to-severe Alzheimer's disease. NEJM. — PubMed
- Rothstein JD et al. (1995). Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Annals of Neurology. — PubMed
- Bensimon G, Lacomblez L, Meininger V (1994). A controlled trial of riluzole in amyotrophic lateral sclerosis. NEJM. — PubMed
- Hardingham GE, Bading H (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nature Reviews Neuroscience. — PubMed
- Nowak L et al. (1984). Magnesium gates glutamate-activated channels in mouse central neurones. Nature. — PubMed
- Mayer ML, Westbrook GL, Guthrie PB (1984). Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. — PubMed
- Iadecola C, Anrather J (2011). The immunology of stroke: from mechanisms to translation. Nature Medicine. — PubMed
PubMed Topic Searches
- PubMed: NMDA receptor and LTP
- PubMed: Glutamate excitotoxicity in stroke
- PubMed: Ketamine for treatment-resistant depression
- PubMed: Memantine for Alzheimer's
- PubMed: Riluzole and ALS
Connections
- Glutamic Acid Benefits Hub
- Glutamic Acid Overview
- GABA Production from Glutamate
- Nitrogen Metabolism
- Glutamate & MSG
- Glutamine (Astrocyte Recycling Partner)
- Glycine (NMDA Co-Agonist)
- Taurine
- Magnesium (Natural NMDA Blocker)
- Vitamin B6 (Glutamate Metabolism Cofactor)
- Epilepsy
- Anxiety
- Migraine
- NAC & Glutathione
- Gut-Brain Axis
- Calcium — the ion that carries the NMDA-receptor signal behind LTP and excitotoxicity.