Glutamic Acid for Nitrogen Metabolism — The Universal Amino Group Carrier
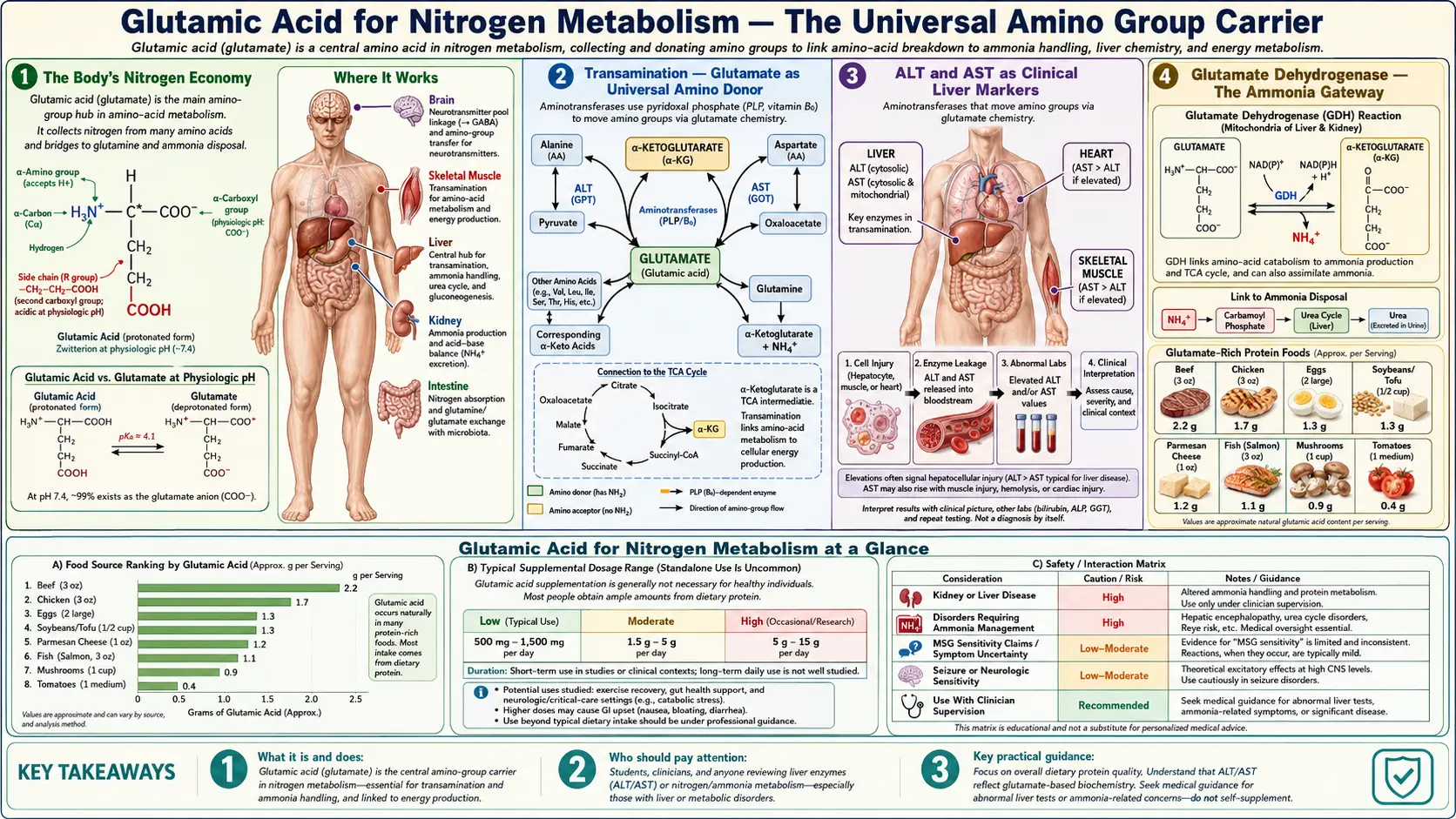
If carbon is the backbone of life, nitrogen is the messenger. Every protein, every nucleotide of DNA and RNA, every essential signaling molecule from neurotransmitters to hormones depends on amino groups derived ultimately from dietary amino acid catabolism — and glutamic acid sits at the absolute center of that flux. Glutamate is the universal amino donor and acceptor in human metabolism: virtually every aminotransferase enzyme uses glutamate / alpha-ketoglutarate as one of its substrate pairs, making glutamate the funnel through which nitrogen passes when amino acids are interconverted, synthesized, or broken down for energy. Glutamate dehydrogenase reversibly couples free ammonia to alpha-ketoglutarate, providing the gateway between organic-bound nitrogen (in amino acids) and inorganic nitrogen (as ammonia or, in the body's waste system, urea). The glutamine-glutamate cycle between astrocytes and neurons in the brain — and a parallel cycle between muscle and liver in the periphery — uses glutamine as the soluble ammonia transport molecule, with glutamate at the heart of every cycle. When the urea cycle fails (genetic enzyme deficiencies, advanced liver disease), hyperammonemia produces hepatic encephalopathy — one of the most severe neurological consequences of disrupted glutamate-mediated nitrogen handling. This deep dive walks through transamination chemistry, glutamate dehydrogenase, the urea cycle entry point, the brain's glutamate-glutamine cycle, hepatic encephalopathy, and the clinical significance for high-protein diets, liver disease, and inborn errors of metabolism.
Table of Contents
- The Body's Nitrogen Economy
- Transamination — Glutamate as Universal Amino Donor
- ALT and AST as Clinical Liver Markers
- Glutamate Dehydrogenase — The Ammonia Gateway
- Urea Cycle Entry and Liver Ammonia Disposal
- The Brain's Glutamate-Glutamine Cycle
- The Muscle-Liver Glutamine and Alanine Cycles
- Hepatic Encephalopathy
- Urea Cycle Disorders
- Alpha-Ketoglutarate as Citric Acid Cycle Intermediate
- Anaplerosis and Cataplerosis
- Clinical Implications for Diet and Disease
- Key Research Papers
- Connections
- Featured Videos
The Body's Nitrogen Economy
The adult human body contains roughly 1.8 to 2.0 kilograms of nitrogen, almost all of it incorporated into proteins, nucleic acids, and small nitrogen-containing molecules like creatine, choline, carnitine, and the porphyrins. Dietary protein supplies the daily nitrogen input: roughly 0.8 grams of protein per kilogram of body weight is the recommended dietary allowance, which translates to about 10-14 grams of nitrogen per day for a 70-kg adult. Excretion of nitrogen as urinary urea balances dietary intake at steady state, with smaller losses through ammonia, creatinine, uric acid, fecal nitrogen, and skin desquamation.
Within the body, nitrogen flows continuously among amino acid pools, protein synthesis and breakdown, neurotransmitter production, nucleic acid turnover, and waste disposal. The flux is enormous — on the order of 300 grams of protein synthesized and broken down per day in an adult (compared to the 60-80 grams ingested), meaning that body protein turns over many times for each gram of dietary protein. This turnover requires extensive amino acid recycling, and that recycling requires extensive nitrogen rearrangement among amino acid carbon skeletons.
Glutamic acid is the molecule that makes that nitrogen rearrangement possible. Through its dual functional groups (an alpha-amino group like every other amino acid, plus a gamma-carboxyl group that allows formation of glutamine, plus the close metabolic relationship to alpha-ketoglutarate as a citric acid cycle intermediate), glutamate sits at the structural and metabolic crossroads where nearly every nitrogen-shuttling pathway meets.
The pithy summary used by biochemistry teachers for decades: "Almost every amino acid in the body has, at some point in its catabolism, passed its amino group to glutamate." It is essentially true.
Transamination — Glutamate as Universal Amino Donor
The reaction that moves amino groups between amino acids is called transamination. The general form:
amino acid1 + alpha-keto acid2 ↔ alpha-keto acid1 + amino acid2
An aminotransferase enzyme catalyzes the transfer of the amino group from amino acid 1 to alpha-keto acid 2, producing the alpha-keto acid of amino acid 1 and the new amino acid 2. The reaction uses pyridoxal 5'-phosphate (PLP) as a covalently bound cofactor through a Schiff-base mechanism — the same chemistry that drives the GAD reaction described on the GABA Production page, but channeled toward amino-group transfer rather than decarboxylation.
What makes glutamate the universal partner is that essentially every aminotransferase uses glutamate / alpha-ketoglutarate as one of its substrate pairs. Examples:
- Alanine aminotransferase (ALT): alanine + alpha-ketoglutarate ↔ pyruvate + glutamate
- Aspartate aminotransferase (AST): aspartate + alpha-ketoglutarate ↔ oxaloacetate + glutamate
- Tyrosine aminotransferase: tyrosine + alpha-ketoglutarate ↔ 4-hydroxyphenylpyruvate + glutamate
- Branched-chain aminotransferase: leucine/isoleucine/valine + alpha-ketoglutarate ↔ alpha-keto acid + glutamate
- Many others — histidine, phenylalanine, ornithine, gamma-aminobutyrate (GABA-T), kynurenine, all funnel their amino groups through transamination with alpha-ketoglutarate to produce glutamate
The result of this universal funneling is that whenever amino acids are catabolized for energy or interconverted, their amino groups end up on glutamate. The body has thus reduced the problem of handling 20+ amino-acid-bound nitrogens to handling a single concentrated pool of glutamate nitrogen — which is then disposed of through glutamate dehydrogenase, glutamine synthesis, or feed-in to the urea cycle. This funnel-and-dispose architecture is one of biology's most elegant pieces of metabolic logic.
ALT and AST as Clinical Liver Markers
The two most commonly measured aminotransferases in clinical practice are alanine aminotransferase (ALT) and aspartate aminotransferase (AST), ordered on essentially every comprehensive metabolic panel and used as the primary serum markers of liver injury. Both are intracellular enzymes that leak into the bloodstream when hepatocytes are damaged.
- ALT is heavily concentrated in the liver and is the more specific marker of hepatocellular injury. Normal range typically 7-55 IU/L. Elevations indicate hepatocyte membrane damage from any cause: viral hepatitis, alcoholic liver disease, non-alcoholic fatty liver disease (NAFLD), drug-induced liver injury, autoimmune hepatitis, ischemia, biliary obstruction.
- AST is more broadly distributed — present in liver, but also heart, skeletal muscle, kidney, brain, and red blood cells. Elevations are less specific. Normal range typically 8-48 IU/L. Heart attack (before troponin took over), muscle injury or rhabdomyolysis, and hemolysis can all elevate AST.
The AST:ALT ratio has additional diagnostic utility. A ratio above 2:1 is classically associated with alcoholic liver disease (mechanism: alcohol depletes pyridoxal phosphate, which is the cofactor for both enzymes, but ALT activity is more sensitive to PLP deficiency than AST, so ALT falls relatively more than AST). A ratio below 1 is more typical of viral hepatitis and most other liver pathologies.
That these two everyday clinical markers are both glutamate-cycling enzymes underscores how central glutamate transamination is to whole-body nitrogen metabolism. When the liver is injured, the leaked aminotransferases reveal themselves — and the fact that these are the enzymes that leak (not, say, the cytochrome P450s or the gluconeogenic enzymes) reflects their extremely high intracellular concentration and their location in the hepatocyte cytoplasm.
Glutamate Dehydrogenase — The Ammonia Gateway
Once all the dietary amino group nitrogens have been funneled onto glutamate via transamination, the body needs a way to liberate that nitrogen as free ammonia for disposal in the urea cycle. The enzyme that performs this critical reaction is glutamate dehydrogenase (GDH), encoded by the GLUD1 gene and concentrated in mitochondria of liver, kidney, intestine, brain, and pancreatic beta cells.
The GDH reaction:
glutamate + NAD(P)+ + H2O ↔ alpha-ketoglutarate + NH3 + NAD(P)H + H+
GDH is unusual in being one of the few mammalian enzymes that uses both NAD+ and NADP+ as cofactors with comparable efficiency. The reaction is freely reversible, allowing GDH to operate either in the oxidative deamination direction (when nitrogen needs to be liberated for urea cycle disposal in the liver) or in the reductive amination direction (when free ammonia needs to be captured into amino acid synthesis — though glutamine synthetase is the higher-affinity enzyme for this purpose).
GDH is intricately regulated. ADP and amino acids (especially leucine) allosterically activate it; GTP, NADH, and palmitoyl-CoA allosterically inhibit it. The leucine activation is the basis of the hyperinsulinism-hyperammonemia (HI/HA) syndrome — a rare autosomal dominant disorder caused by gain-of-function mutations in GLUD1. Affected children have constitutively over-active GDH, which produces both excessive alpha-ketoglutarate (driving insulin secretion in pancreatic beta cells and causing hypoglycemia, especially after protein meals) and excessive ammonia (causing modest chronic hyperammonemia). Treatment includes diazoxide for the hypoglycemia and dietary protein management.
In the brain, GDH activity is one of the limiting steps in the metabolism of glutamate to alpha-ketoglutarate as a TCA cycle anaplerotic source. Deficient GDH activity in the brain (genetic or pharmacological) has been linked to certain forms of epilepsy and cognitive impairment.
Urea Cycle Entry and Liver Ammonia Disposal
Free ammonia is highly toxic at concentrations above approximately 50 micromolar (normal plasma ammonia is <35 micromolar). The terrestrial mammalian solution to disposing of nitrogen waste is the urea cycle, which converts two ammonia molecules and one carbon dioxide into one molecule of urea, which is non-toxic, water-soluble, and excreted in urine. The urea cycle takes place exclusively in liver hepatocytes (a small amount in kidney for arginine biosynthesis), and it depends critically on glutamate-mediated ammonia delivery.
The urea cycle has five enzymatic steps, of which the first is the rate-limiting:
- Carbamoyl phosphate synthetase I (CPS1) — mitochondrial, condenses ammonia, bicarbonate, and 2 ATP to form carbamoyl phosphate. Requires N-acetylglutamate (NAG) as an absolute allosteric activator. NAG is synthesized from glutamate + acetyl-CoA by N-acetylglutamate synthase (NAGS) — another point where glutamate sits at a critical regulatory junction.
- Ornithine transcarbamylase (OTC) — mitochondrial, transfers carbamoyl group to ornithine to make citrulline. OTC deficiency is the most common urea cycle disorder, X-linked, presenting in males with hyperammonemic coma in early infancy.
- Argininosuccinate synthetase (ASS) — cytoplasmic, joins citrulline with aspartate (which carries the second urea nitrogen) to form argininosuccinate. The aspartate is supplied by transamination from glutamate — another glutamate input.
- Argininosuccinate lyase (ASL) — cytoplasmic, cleaves argininosuccinate to arginine plus fumarate. The fumarate enters the citric acid cycle, providing the link that allows urea cycle nitrogen disposal to be coupled to oxidative metabolism.
- Arginase — cytoplasmic, hydrolyzes arginine to urea plus ornithine. The ornithine returns to the mitochondrion for the next turn of the cycle.
Glutamate feeds the urea cycle in three distinct ways: (1) as the source of N-acetylglutamate that allosterically activates CPS1; (2) as the source of free ammonia via GDH for the first urea nitrogen; and (3) as the source of aspartate (via AST transamination of oxaloacetate) for the second urea nitrogen. The cycle would not run without glutamate.
The carbamoyl phosphate synthetase II (CPS2) used in pyrimidine biosynthesis, by contrast, uses glutamine as its nitrogen donor and is regulated entirely differently. This distinction (CPS1 for urea, CPS2 for pyrimidines) trips up many medical students.
The Brain's Glutamate-Glutamine Cycle
The brain manufactures essentially all of its own glutamate because the blood-brain barrier severely restricts glutamate uptake from the periphery. The carbon backbone comes from glucose-derived alpha-ketoglutarate via the citric acid cycle. The amino group comes ultimately from ammonia or from amino acid catabolism. The cycling between glutamate and glutamine that connects neurons and astrocytes was introduced on the Neurotransmission page; here we look at it from the nitrogen-balance perspective.
The brain's glutamine synthetase activity is enormous — concentrated in astrocytes, it efficiently captures both the glutamate released at synapses and any ammonia generated locally. The reaction:
glutamate + NH3 + ATP → glutamine + ADP + Pi
The glutamine product is non-toxic (unlike free glutamate, which is excitotoxic, and unlike free ammonia, which is neurotoxic) and freely diffusible. Astrocytic glutamine is exported across the blood-brain barrier into the systemic circulation, where it serves as the major soluble nitrogen carrier in the body. Glutamine concentration in plasma is approximately 600-700 micromolar — the highest of any amino acid — reflecting its dominant role in inter-organ nitrogen shuttling.
When peripheral ammonia rises (as in liver failure), the brain's glutamine synthetase keeps churning, but in reverse logic — it has to deal with the influx of ammonia crossing the BBB. The result is intracellular astrocytic glutamine accumulation that draws water osmotically into astrocytes, causing them to swell and contributing to the cerebral edema of hepatic encephalopathy and ammonia toxicity.
The Muscle-Liver Glutamine and Alanine Cycles
The body has two parallel inter-organ nitrogen-shuttling cycles, both centered on glutamate:
- The glutamine cycle — skeletal muscle, brain, lung, and adipose tissue release glutamine into the bloodstream. Glutamine is the most abundant amino acid in plasma at 600-700 micromolar. The liver, kidney, gut, and rapidly dividing cells (lymphocytes, enterocytes) take up glutamine and deaminate it (releasing the nitrogen for urea or new amino acid synthesis) and use the alpha-ketoglutarate carbon skeleton for energy or biosynthesis. Glutamine is so important to gut and immune cells that under critical illness conditions, it becomes conditionally essential — the body cannot synthesize enough to meet demand, and supplementation may be beneficial. See the Glutamine page for details.
- The alanine cycle (Cahill cycle) — named after George Cahill who described it, this cycle shuttles nitrogen and carbon between exercising muscle and liver in a manner analogous to the Cori cycle for lactate. Muscle takes glucose, converts it to pyruvate via glycolysis, transaminates pyruvate to alanine using ALT (donating the amino group from branched-chain amino acid catabolism via glutamate), releases alanine into blood, which the liver takes up. Liver ALT reverses the reaction (alanine + alpha-ketoglutarate → pyruvate + glutamate), uses pyruvate for gluconeogenesis (returning glucose to muscle to complete the loop), and feeds the glutamate nitrogen into the urea cycle. The alanine cycle is one of the major routes by which muscle protein catabolism during fasting or prolonged exercise contributes to hepatic gluconeogenesis.
Both cycles depend on glutamate / alpha-ketoglutarate cycling. In the muscle-liver alanine cycle, glutamate is the immediate carrier of branched-chain amino acid nitrogen from muscle catabolism to alanine. In the broader glutamine cycle, glutamate is the precursor for glutamine synthesis (via glutamine synthetase) in muscle and the product of glutamine deamination (via phosphate-activated glutaminase) in liver.
Hepatic Encephalopathy
When the liver fails — either acutely (acute liver failure from acetaminophen overdose, viral hepatitis, ischemia) or chronically (cirrhosis with portosystemic shunting) — the urea cycle cannot keep up with nitrogen disposal demand. Ammonia accumulates in the blood, crosses the blood-brain barrier, and disrupts brain function. The clinical syndrome is hepatic encephalopathy, which ranges from minimal cognitive impairment detectable only on neuropsychological testing to severe disorientation, asterixis (the classic flapping tremor), stupor, and coma.
The neurochemistry of hepatic encephalopathy centers on glutamate. The excess ammonia is taken up by astrocytes and used by glutamine synthetase to make glutamine. The glutamine accumulates intracellularly, draws water osmotically into the astrocyte, and causes astrocyte swelling. The swelling produces cerebral edema (most severe in acute liver failure), and the resulting changes in neuronal-astrocyte coupling alter glutamatergic neurotransmission, GABAergic neurotransmission (often paradoxically enhanced, possibly explaining the sedation of HE), and overall brain energy metabolism.
The clinical management of HE is mechanistically aimed at reducing ammonia load:
- Lactulose — non-absorbable disaccharide that acidifies the colon, trapping ammonia as ammonium ion for fecal excretion and shifting gut microbiome composition. First-line therapy.
- Rifaximin — minimally absorbed antibiotic that reduces gut bacterial ammonia production. Often added to lactulose for recurrent HE.
- L-ornithine L-aspartate (LOLA) — provides substrates for urea cycle and glutamine synthesis. Used in some countries.
- Branched-chain amino acid supplementation — replaces aromatic amino acids that may contribute to false neurotransmitter formation in HE.
- Protein restriction — historically advocated, now largely abandoned because protein restriction worsens sarcopenia and overall outcomes. Adequate protein intake (1.2-1.5 g/kg/day) with vegetable and dairy sources preferred over red meat for any potential ammonia advantage.
- Liver transplantation — the definitive treatment for HE in cirrhosis.
HE is one of the clearest demonstrations that brain function depends on intact systemic nitrogen handling — and that intact systemic nitrogen handling depends on glutamate biology.
Urea Cycle Disorders
Inherited defects in any of the five urea cycle enzymes (or the NAGS regulator) produce hyperammonemia with a clinical phenotype that ranges from neonatal catastrophic coma (severe deficiencies) to adult-onset behavior changes and recurrent vomiting episodes (partial deficiencies). The order of incidence:
- Ornithine transcarbamylase (OTC) deficiency — X-linked, most common urea cycle disorder. Affected males present in neonatal period with coma after first protein feedings. Heterozygous females have variable presentation depending on X-inactivation patterns, sometimes mild and not recognized until adulthood.
- Argininosuccinate synthetase (ASS) deficiency — citrullinemia type I. Autosomal recessive. Plasma citrulline is greatly elevated.
- Argininosuccinate lyase (ASL) deficiency — argininosuccinic aciduria. Autosomal recessive. Trichorrhexis nodosa (brittle hair) is a classic feature.
- Carbamoyl phosphate synthetase I (CPS1) deficiency — autosomal recessive, severe neonatal presentation.
- N-acetylglutamate synthase (NAGS) deficiency — very rare; the only urea cycle disorder for which a specific drug (carglumic acid, the synthetic NAG analog) provides essentially complete biochemical correction.
- Arginase deficiency — argininemia. Unusual clinical phenotype: spastic paraparesis and progressive intellectual disability rather than the typical hyperammonemic crises.
Management of urea cycle disorders involves dietary protein restriction (carefully balanced to provide enough essential amino acids without overloading the broken cycle), nitrogen scavenger drugs (sodium phenylacetate / sodium benzoate, which conjugate with glutamine and glycine respectively to form excreted nitrogen waste products that bypass urea), and emergency hemodialysis for severe hyperammonemia. Liver transplantation provides definitive cure for severe forms.
Recent advances include mRNA therapeutics, AAV-mediated gene therapy (particularly for OTC deficiency), and the small-molecule chaperone glycerol phenylbutyrate for ammonia management.
Alpha-Ketoglutarate as Citric Acid Cycle Intermediate
Glutamate's carbon skeleton, alpha-ketoglutarate, is one of the eight intermediates of the citric acid cycle (Krebs cycle, TCA cycle). This dual identity — nitrogen carrier as glutamate, energy intermediate as alpha-ketoglutarate — is what links amino acid metabolism so tightly to central energy metabolism.
Alpha-ketoglutarate (also written as 2-oxoglutarate) sits between isocitrate and succinyl-CoA in the cycle. The forward direction (isocitrate → alpha-ketoglutarate → succinyl-CoA) is catabolic, generating CO2, NADH, and ATP-equivalent reducing power. The conversion alpha-ketoglutarate → succinyl-CoA is catalyzed by alpha-ketoglutarate dehydrogenase — a thiamine-dependent enzyme complex analogous to pyruvate dehydrogenase, and one of the most sensitive enzymes in the cell to oxidative damage (reduced alpha-KG dehydrogenase activity is one of the earliest biochemical changes in Alzheimer's disease and Parkinson's disease brain tissue).
Alpha-ketoglutarate also serves several non-energy roles:
- Cofactor for prolyl hydroxylase — the enzyme that hydroxylates HIF-1alpha (hypoxia-inducible factor) under normoxia, marking it for degradation. Under hypoxia, when alpha-KG is depleted or when oxygen runs short, prolyl hydroxylase activity falls, HIF-1alpha accumulates, and the cell launches its hypoxia-response program (erythropoietin, VEGF, glycolytic enzymes). This is the basis for the prolyl hydroxylase inhibitors (roxadustat and others) used for anemia of chronic kidney disease.
- Cofactor for TET enzymes — ten-eleven translocation methylcytosine dioxygenases that demethylate DNA at CpG sites, a key epigenetic regulation step. TET enzymes are highly sensitive to the alpha-KG / 2-hydroxyglutarate ratio.
- Cofactor for collagen hydroxylation — proline and lysine hydroxylation in nascent collagen chains requires alpha-KG, ascorbate, and oxygen. This is one of the mechanisms underpinning vitamin C's role in collagen synthesis and the scurvy phenotype.
- Anti-aging research target — alpha-ketoglutarate supplementation has been investigated for longevity effects with promising animal-model data and limited but encouraging human research.
The cancer connection: mutations in the metabolic enzymes IDH1 and IDH2 (isocitrate dehydrogenase 1 and 2), found in many gliomas and acute myeloid leukemias, cause aberrant production of 2-hydroxyglutarate instead of alpha-ketoglutarate. 2-HG is a competitive inhibitor of alpha-KG-dependent enzymes including the TET DNA demethylases, producing the characteristic CpG island methylator phenotype of IDH-mutant tumors. The targeted IDH inhibitors (ivosidenib for IDH1, enasidenib for IDH2) restore normal alpha-KG production and reverse the epigenetic perturbations.
Anaplerosis and Cataplerosis
Two technical terms describe the way glutamate / alpha-ketoglutarate cycling affects citric acid cycle flux:
- Anaplerosis (from Greek "filling up") — the addition of carbon skeletons to the citric acid cycle from external sources. Glutamate → alpha-ketoglutarate via GDH or transamination is a major anaplerotic input, particularly in the brain and in tumors with high glutamine consumption. Other anaplerotic reactions include pyruvate carboxylase (pyruvate → oxaloacetate) and propionyl-CoA carboxylase (from odd-chain fatty acid and BCAA catabolism). Anaplerosis is needed because cycle intermediates are constantly being siphoned off for biosynthesis (e.g., citrate → fatty acid synthesis, alpha-KG → glutamate, oxaloacetate → gluconeogenesis), and without replacement the cycle would deplete and grind to a halt.
- Cataplerosis — the removal of carbon skeletons from the citric acid cycle for biosynthesis. Alpha-ketoglutarate → glutamate (via reverse GDH or transamination) is the major cataplerotic exit from the cycle for amino acid synthesis. Citrate → cytoplasmic acetyl-CoA for fatty acid and cholesterol synthesis is another major cataplerotic flux.
In rapidly dividing cells (cancer, immune cells, gut epithelium), the demand for both anaplerotic input (to replace siphoned carbons) and cataplerotic output (for biosynthesis) is high — which is why many cancer cells become "glutamine addicted," consuming enormous quantities of glutamine through glutaminase and GDH to maintain cycle flux for biosynthesis. The glutaminase inhibitor CB-839 (telaglenastat) has been in clinical trials for cancers exhibiting this dependency.
The bidirectional flexibility of glutamate / alpha-KG — serving as either anaplerotic input or cataplerotic output as cellular demands shift — is what makes it the central metabolic hub of intermediary metabolism.
Clinical Implications for Diet and Disease
The central nitrogen-handling role of glutamate has practical implications for several common clinical situations:
- High-protein diets — ketogenic, carnivore, and high-protein weight-loss diets impose substantially elevated demands on urea cycle flux. Healthy livers handle this without difficulty (humans evolved on intermittent high-protein meat consumption), but patients with subclinical liver dysfunction, partial urea cycle enzyme deficiencies (heterozygous OTC carriers, for example), or chronic alcohol abuse may experience ammonia symptoms (brain fog, fatigue, headache) on protein loads they previously tolerated.
- Cirrhosis and chronic liver disease — protein intake is best maintained at 1.2-1.5 g/kg/day (higher than general adult RDA) to prevent sarcopenia, with vegetable and dairy proteins preferred over red meat for the slight ammonia advantage. Branched-chain amino acid supplementation may help. Lactulose for prevention of recurrent HE.
- Endurance exercise and athletic recovery — prolonged exercise increases skeletal muscle glutamine release and the alanine cycle flux. Glutamine becomes conditionally essential under heavy training loads. Adequate carbohydrate intake spares amino acid catabolism. See the Glutamine page.
- Critical illness, surgery, burns, sepsis — massive protein catabolism, glutamine and arginine become conditionally essential. Glutamine supplementation in select ICU populations has been beneficial in some trials, though high-dose IV glutamine in some critically ill populations was associated with increased mortality in the REDOXS trial — the picture is nuanced and clinical situation-specific.
- Inborn errors of metabolism — urea cycle disorders, MSUD (maple syrup urine disease, BCAA catabolism), propionic and methylmalonic acidemias, and the organic acidemias all impinge on glutamate nitrogen flow. Specialty metabolic genetics consultation is essential.
- Vitamin B6 status — because every transaminase and the GAD reaction depend on PLP, B6 status directly affects whole-body nitrogen flexibility. Adequate intake (1.3-2.0 mg/day food + supplements) is the foundation; higher therapeutic doses (25-100 mg/day P5P) for specific deficiencies under guidance. See the Vitamin B6 page.
- Liver-supportive botanicals — milk thistle, dandelion root, artichoke leaf have evidence for hepatocyte protection and improved bile flow. Adequate water intake and avoiding hepatotoxic substances (excess alcohol, acetaminophen above recommended doses, isolated high-dose niacin) is foundational.
The takeaway: glutamate's role as the universal nitrogen carrier of human metabolism means that virtually every clinical situation involving protein nutrition, liver function, neurotransmitter balance, or energy metabolism is touched by glutamate biology. Supporting adequate B6, magnesium, and overall mitochondrial function provides the metabolic foundation for healthy glutamate-mediated nitrogen flow.
Key Research Papers
- Krebs HA, Henseleit K (1932). Untersuchungen über die Harnstoffbildung im Tierkörper. Klinische Wochenschrift. — PubMed
- Cooper AJ, Plum F (1987). Biochemistry and physiology of brain ammonia. Physiological Reviews. — PubMed
- Stanley CA et al. (1998). Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. NEJM. — PubMed
- Felig P (1973). The glucose-alanine cycle. Metabolism. — PubMed
- Newsholme P et al. (2003). Glutamine and glutamate as vital metabolites. Brazilian Journal of Medical and Biological Research. — PubMed
- Haussinger D et al. (2000). Hepatic encephalopathy in chronic liver disease: a clinical manifestation of astrocyte swelling and low-grade cerebral edema? Journal of Hepatology. — PubMed
- Tuchman M, Caldovic L et al. (2008). N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate synthase deficiency. Pediatric Research. — PubMed
- Ward LC (2017). Branched-chain amino acid metabolism in nutritional support of the critically ill. Critical Care. — PubMed
- Wise DR, Thompson CB (2010). Glutamine addiction: a new therapeutic target in cancer. Trends in Biochemical Sciences. — PubMed
- Dang L et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. — PubMed
- Heird WC (1998). Amino acids in pediatric and neonatal nutrition. Current Opinion in Clinical Nutrition and Metabolic Care. — PubMed
- Heyland D et al. (2013). A randomized trial of glutamine and antioxidants in critically ill patients (REDOXS). NEJM. — PubMed
PubMed Topic Searches
- PubMed: Glutamate dehydrogenase and ammonia
- PubMed: Urea cycle disorders
- PubMed: Hepatic encephalopathy
- PubMed: Glutamate-glutamine cycle
- PubMed: ALT/AST and liver disease
Connections
- Glutamic Acid Benefits Hub
- Glutamic Acid Overview
- Neurotransmission
- GABA Production
- Glutamate & MSG
- Glutamine (Nitrogen Transport)
- Alanine (Alanine Cycle Partner)
- Aspartic Acid (Second Urea Nitrogen)
- Arginine (Urea Cycle)
- Vitamin B6 (Transamination Cofactor)
- Cirrhosis & Liver Disease
- Lab Tests (ALT/AST)
- Proline (Glutamate Precursor)
- NAC & Glutathione
- Milk Thistle (Liver Support)