Arginine for Cardiovascular Health and Nitric Oxide
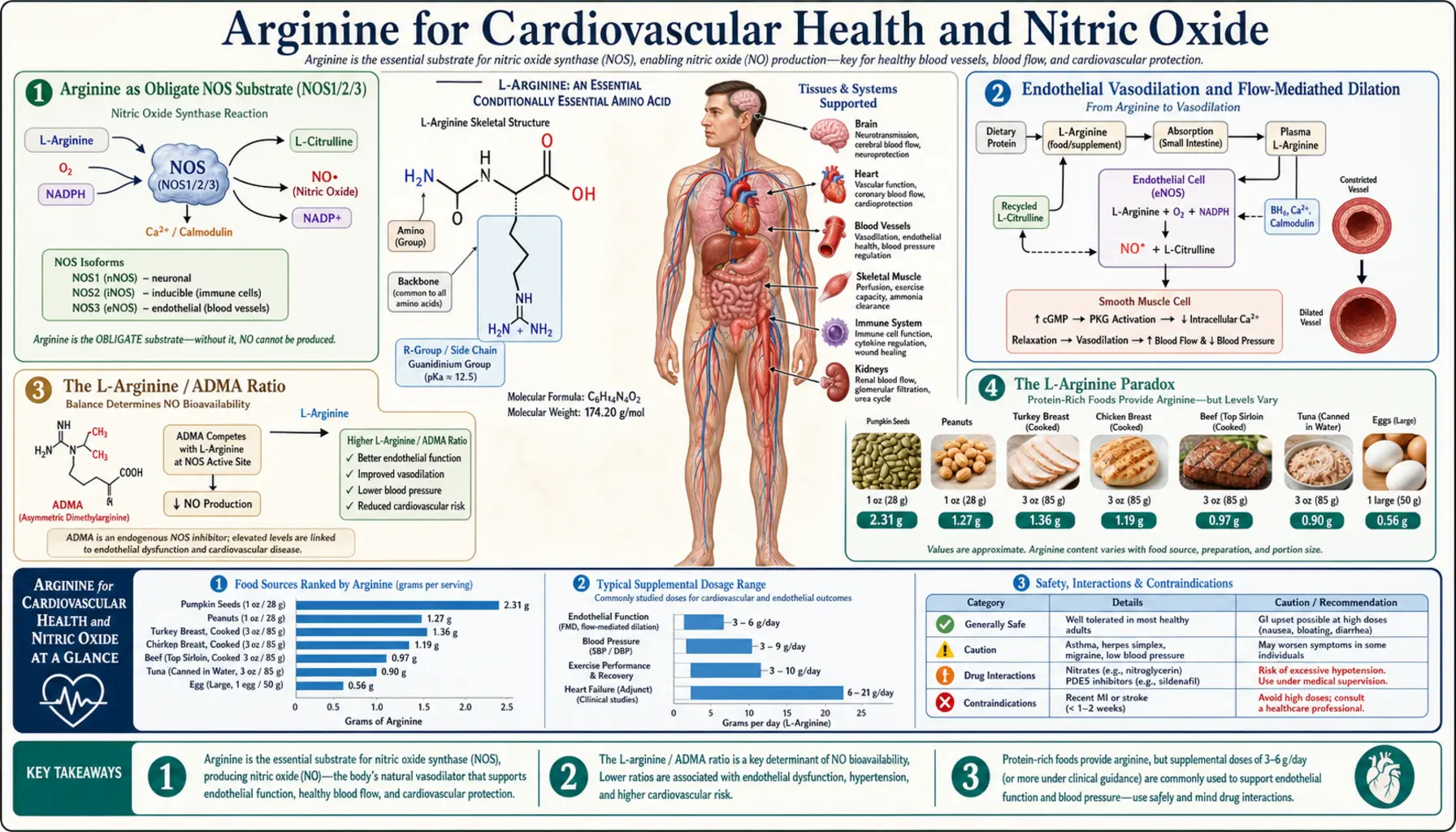
L-Arginine is the obligate physiological substrate for the entire nitric oxide synthase (NOS) family — the three isoforms NOS1 (neuronal), NOS2 (inducible), and NOS3 (endothelial) all draw on the same intracellular arginine pool to generate the gaseous signaling molecule that relaxes vascular smooth muscle, prevents platelet aggregation, suppresses leukocyte adhesion, and underwrites the integrity of the endothelium. Endothelial dysfunction — defined operationally as impaired flow-mediated dilation — is the earliest detectable abnormality of atherosclerosis, often present a decade or more before the first chest pain. The arginine-NO axis sits at the dead center of that vascular biology. Yet the clinical translation has been complicated: the L-arginine paradox in heart failure, the negative VINTAGE-MI trial in post-infarct patients, and the unexpected finding that citrulline is often a more bioavailable NO precursor than arginine itself together make this one of the most scientifically interesting and clinically nuanced supplement stories in cardiovascular medicine. This deep-dive walks through the biochemistry, the trial evidence, and the practical implications for patients with hypertension, endothelial dysfunction, peripheral arterial disease, and stable angina.
Table of Contents
- Arginine as Obligate NOS Substrate (NOS1/2/3)
- Endothelial Vasodilation and Flow-Mediated Dilation
- The L-Arginine / ADMA Ratio
- The L-Arginine Paradox
- Citrulline as a More Bioavailable NO Precursor
- Hepatic Arginase and First-Pass Loss
- Hypertension Trial Evidence
- Peripheral Arterial Disease and Stable Angina
- Heart Failure and the VINTAGE-MI Trial
- Clinical Practice Recommendations
- Cautions and Contraindications
- Key Research Papers
- Connections
- Featured Videos
Arginine as Obligate NOS Substrate (NOS1/2/3)
Nitric oxide synthase is the enzyme family that converts L-arginine to L-citrulline and nitric oxide, consuming one molecule of NADPH and one molecule of O2 per turnover. The reaction is mechanistically a five-electron oxidation of the guanidino nitrogen of arginine, and arginine is the only physiological substrate that the active site will accept — no other amino acid, no shortcut, no alternative pathway. If intracellular arginine drops below the Michaelis constant of the enzyme (approximately 3 µM for NOS3), nitric oxide production drops proportionally.
Three NOS isoforms are encoded by distinct genes on different chromosomes and serve distinct functions:
- NOS1 (neuronal NOS, nNOS) — constitutively expressed, calcium/calmodulin-dependent, located in central and peripheral neurons. Produces NO that functions as a non-classical neurotransmitter regulating long-term potentiation, gastrointestinal smooth muscle relaxation, and penile erection.
- NOS2 (inducible NOS, iNOS) — not constitutively expressed; induced by inflammatory cytokines (interferon-gamma, TNF-alpha, IL-1) in macrophages, neutrophils, and other cells. Calcium-independent, produces large sustained bursts of NO at micromolar concentrations as a cytotoxic effector against bacteria, parasites, and tumor cells.
- NOS3 (endothelial NOS, eNOS) — constitutively expressed in vascular endothelial cells, calcium/calmodulin-dependent, activated by shear stress, bradykinin, acetylcholine, and other endothelium-dependent vasodilators. Produces NO at nanomolar concentrations that diffuses into adjacent vascular smooth muscle, activates soluble guanylate cyclase, generates cGMP, and triggers vasorelaxation.
The substrate dependency is absolute for all three. Pharmacological NOS inhibitors such as L-NMMA (N-monomethyl-L-arginine) and L-NAME (N-omega-nitro-L-arginine methyl ester) work by competing with arginine for the active site — the proof of concept that arginine availability is the rate-limiting input to NO synthesis under most physiological conditions. The implication is that any clinical state that depletes intracellular arginine, or any genetic or acquired condition that elevates competitive NOS inhibitors like asymmetric dimethylarginine (ADMA), will measurably reduce endothelial NO production and the downstream vascular effects.
Endothelial Vasodilation and Flow-Mediated Dilation
The vascular endothelium is not the inert lining once described in 19th-century anatomy textbooks — it is a continuous monolayer of approximately one trillion cells with a combined surface area equivalent to a tennis court, and it is metabolically one of the most active tissues in the body. Endothelial cells continuously sense mechanical shear stress from flowing blood, biochemical signals from circulating hormones and platelets, and local cytokines from adjacent immune cells. The endothelium translates these inputs into the release of vasoactive mediators that control vascular tone, platelet behavior, leukocyte trafficking, and smooth muscle phenotype.
Nitric oxide is the most important of these mediators. Released continuously at low basal levels by NOS3, NO maintains a tonic vasodilator influence that opposes the constrictor influence of sympathetic catecholamines, angiotensin II, and endothelin-1. The clinical gold standard for measuring endothelial NO function is flow-mediated dilation (FMD) of the brachial artery: after five minutes of distal forearm occlusion with a blood pressure cuff, the released hyperemic blood flow generates increased shear stress on the brachial endothelium, which activates NOS3 and dilates the artery. The dilation is measured by high-resolution ultrasound and expressed as a percent increase from baseline diameter. Healthy young adults dilate 8–12%; older adults with cardiovascular risk factors dilate 3–5%; patients with established coronary disease often dilate less than 2%.
Multiple randomized controlled trials have demonstrated that oral L-arginine improves FMD in patients with documented endothelial dysfunction. The effect size is modest but consistent — typically a 1–2 percentage-point absolute improvement in FMD, sustained over weeks of supplementation at 3–6 g/day. The improvement is most pronounced in patients with hypercholesterolemia, diabetes, smoking history, hypertension, or other conditions associated with elevated ADMA. Patients with normal baseline endothelial function show no measurable benefit, consistent with the view that arginine repletion corrects a relative deficit rather than enhancing already-adequate substrate supply.
The vascular consequences of restored endothelial function extend beyond simple vasodilation. Improved NO bioavailability also reduces platelet aggregation (NO activates platelet guanylate cyclase, raising platelet cGMP, which inhibits aggregation), suppresses leukocyte adhesion to endothelium (NO downregulates VCAM-1 and ICAM-1 expression), and reduces vascular smooth muscle cell proliferation (NO inhibits PDGF-induced mitogenesis). The net effect is anti-atherogenic across multiple pathophysiologic axes.
The L-Arginine / ADMA Ratio
Asymmetric dimethylarginine (ADMA) is a naturally occurring methylated derivative of arginine, produced when methylated arginine residues in proteins are released during normal protein turnover. ADMA is structurally similar enough to arginine to bind the NOS active site, but it cannot be turned over to NO — it is a competitive inhibitor. Under normal conditions, ADMA is degraded by the enzyme dimethylarginine dimethylaminohydrolase (DDAH) to citrulline and dimethylamine, keeping plasma ADMA in the 0.4–0.6 µmol/L range.
ADMA rises in clinical states characterized by oxidative stress, because DDAH is itself inhibited by reactive oxygen species. Elevated ADMA is found in chronic kidney disease (where renal clearance is impaired and oxidative stress is elevated), diabetes, hypertension, hyperhomocysteinemia, hypercholesterolemia, smoking, aging, and pregnancy complications (preeclampsia). Plasma ADMA above 0.7 µmol/L is associated with approximately 2-fold increased cardiovascular mortality risk in multiple cohort studies, including the AtheroGene study and the Framingham Offspring Study.
The key insight is that the absolute concentration of ADMA matters less than the L-arginine to ADMA ratio. NOS sees a competition at its active site, and the outcome depends on the relative concentrations of substrate (arginine) and inhibitor (ADMA). A patient with normal arginine but elevated ADMA can have severely impaired NO production; the same patient with arginine boosted to the upper end of normal can effectively out-compete the ADMA and restore NO production.
This is one of the strongest mechanistic rationales for arginine supplementation in clinical practice. Patients with elevated ADMA — readily measured by HPLC or LC-MS in specialty labs — are the population most likely to respond. The arginine/ADMA ratio is increasingly used as a clinical biomarker for endothelial dysfunction and is part of several emerging cardiovascular risk-stratification scores in research settings.
The L-Arginine Paradox
The L-arginine paradox is the observation that exogenous arginine increases NOS-mediated NO production despite the fact that intracellular arginine is already present at concentrations far exceeding the Michaelis constant of NOS3 (the Km of approximately 3 µM is dwarfed by intracellular arginine concentrations of 100–800 µM). By standard enzyme kinetics, the enzyme should already be operating at Vmax, and adding more substrate should produce no additional product. Yet decades of human studies clearly show that oral arginine raises NO production in dysfunctional endothelium.
Several mechanisms have been proposed to explain this apparent contradiction:
- Compartmentalization — the arginine pool seen by NOS is not the same as the cytosolic bulk arginine measured in cell lysates. NOS3 is anchored in caveolae (specialized membrane microdomains) and shares this compartment with the cationic amino acid transporter CAT-1, which delivers arginine across the plasma membrane directly to the enzyme. The caveolar arginine concentration may be far lower than bulk cytosolic arginine, and exogenous arginine may preferentially fill this functional pool.
- ADMA competition — as discussed above, the relevant kinetic parameter is the arginine/ADMA ratio, not absolute arginine concentration. Adding arginine shifts the ratio in favor of substrate and out-competes the inhibitor.
- BH4 cofactor coupling — NOS requires tetrahydrobiopterin (BH4) as an essential cofactor. Under oxidative stress, BH4 is oxidized to BH2, and BH2-bound NOS becomes "uncoupled" — it produces superoxide instead of NO. The uncoupling worsens oxidative stress in a vicious cycle. Arginine supplementation may favor proper substrate-cofactor coupling and reduce superoxide generation by uncoupled NOS.
- Arginase competition — in some clinical states (asthma, sickle cell disease, certain cancers, advanced age), the arginase enzyme is upregulated and consumes arginine via the ornithine/urea cycle pathway, depleting the substrate pool available to NOS. Exogenous arginine compensates for this depletion.
The paradox is therefore not actually a paradox — it is a clinical signal that the simple textbook model of NOS enzymology fails to capture the relevant biology. In dysfunctional endothelium, multiple mechanisms simultaneously limit NO production, and supplementing the substrate addresses several of them at once.
Citrulline as a More Bioavailable NO Precursor
One of the most important practical developments in the arginine-NO field over the past two decades is the recognition that L-citrulline, the byproduct of the NOS reaction itself, is often a more effective oral NO precursor than L-arginine. Citrulline is converted back to arginine in the kidneys by the two-enzyme sequence argininosuccinate synthetase (ASS) and argininosuccinate lyase (ASL), and this conversion serves as a continuous endogenous arginine supply.
Critically, citrulline is not subject to first-pass hepatic metabolism by arginase. Hepatic arginase is the dominant enzyme of the urea cycle, present at extremely high concentration in periportal hepatocytes, and it efficiently consumes any arginine delivered via the portal circulation, converting it to ornithine and urea. The result is that approximately 40% of an oral arginine dose is destroyed by first-pass hepatic arginase before it reaches the systemic circulation. Oral citrulline bypasses this loss entirely — it is absorbed intact, passes through the liver unmolested, and is converted to arginine in the proximal tubule cells of the kidney, from which it enters the systemic arginine pool.
The kinetic consequence is that an oral dose of citrulline produces a higher and more sustained plasma arginine concentration than the same molar dose of arginine itself. Pharmacokinetic studies by Schwedhelm et al. and Moinard et al. have shown that 3 g of oral citrulline produces plasma arginine levels approximately equivalent to 6 g of oral arginine, and the citrulline-driven rise persists for 6–8 hours versus 1–2 hours for direct arginine. Citrulline is also better tolerated — the high osmotic load of oral arginine often causes gastrointestinal distress (nausea, cramping, diarrhea) at doses above 5–10 g, while citrulline is well-tolerated at doses up to 15 g.
Clinical trials directly comparing the two have generally favored citrulline. In the trial by Suzuki et al. (2016), 1.2 g/day of citrulline produced better flow-mediated dilation improvement than the same dose of arginine in patients with cardiovascular risk factors. In the trial by Bailey et al. (2015) in endurance cyclists, 6 g of citrulline produced larger increases in exercise capacity than 6 g of arginine. In erectile dysfunction studies, citrulline has produced symptomatic benefit at doses (1.5 g/day) that are far below the effective arginine doses (3–5 g/day).
For most cardiovascular applications, citrulline has become the preferred NO precursor in clinical practice, especially in patients sensitive to the GI side effects of oral arginine. The exception is the WHO measles-style scenario where rapid and massive NOS substrate loading is the goal (intravenous arginine, used in some critical care protocols), but for outpatient daily supplementation, citrulline is biochemically superior.
Hepatic Arginase and First-Pass Loss
Arginase is the terminal enzyme of the urea cycle. It hydrolyzes arginine to ornithine and urea, the latter being excreted as the principal nitrogenous waste product in mammals. There are two arginase isoforms encoded by separate genes:
- Arginase 1 (ARG1) — cytosolic, expressed at extremely high concentration in periportal hepatocytes (liver) and at lower levels in red blood cells, certain immune cells, and the inner medulla of the kidney. Functions in the urea cycle to detoxify ammonia from amino acid catabolism. The hepatic concentration is so high that ARG1 is one of the most abundant proteins in liver cytosol.
- Arginase 2 (ARG2) — mitochondrial, expressed widely in kidney, prostate, small intestine, lactating mammary gland, and macrophages. Lower abundance than ARG1 but more widely distributed. Functions in ornithine and polyamine production rather than primary urea generation.
The clinical relevance of hepatic ARG1 is that it directly competes with NOS for the same substrate. Any oral arginine dose passes through the portal vein, traverses the liver, and is exposed to periportal ARG1 before reaching the systemic circulation. The first-pass loss is substantial — estimates from stable-isotope tracer studies in humans range from 30–50% of the oral dose. The remaining 50–70% that reaches systemic circulation can be further consumed by extrahepatic arginase isoforms before reaching endothelial NOS.
This first-pass loss becomes clinically important in conditions that upregulate arginase activity:
- Asthma — airway epithelial arginase is induced by Th2 cytokines (IL-4, IL-13), depleting arginine in the bronchial mucosa and contributing to airway hyperresponsiveness
- Sickle cell disease — intravascular hemolysis releases erythrocyte arginase 1 into plasma, where it depletes circulating arginine. This contributes to the pulmonary hypertension and vaso-occlusive crises characteristic of the disease
- Sepsis — circulating arginase activity rises dramatically in septic patients, producing functional arginine deficiency and contributing to the microvascular dysfunction of septic shock
- Heart failure — arginase activity is upregulated in failing myocardium and contributes to NOS uncoupling and oxidative stress
- Cancer (myeloid-derived suppressor cells) — tumor-associated MDSCs produce arginase to deplete arginine in the tumor microenvironment, suppressing T-cell function (discussed further on the Immune Function page)
In all of these conditions, the arginase upregulation creates a state of functional arginine deficiency that conventional plasma arginine measurement may underestimate. Citrulline supplementation, by bypassing hepatic arginase entirely, is particularly advantageous in these patients.
Hypertension Trial Evidence
The clinical trial evidence for arginine in hypertension is positive but modest in effect size. The most-cited meta-analysis is by Dong et al. (2011), which pooled 11 randomized controlled trials of oral L-arginine versus placebo in adults with elevated blood pressure. The pooled effect was a reduction of approximately 5.4 mmHg in systolic blood pressure and 2.7 mmHg in diastolic blood pressure, both statistically significant. The effect was dose-dependent up to about 9 g/day; higher doses did not produce proportionally larger reductions.
For context, these effect sizes are roughly half the magnitude of a typical low-dose antihypertensive medication (a starting dose of an ACE inhibitor or thiazide diuretic produces approximately 10–12 mmHg systolic reduction in unselected hypertensive patients). Arginine is therefore not a first-line antihypertensive substitute, but it can be a useful adjunct, particularly in patients with documented endothelial dysfunction, elevated ADMA, or hypertension associated with specific conditions:
- Gestational hypertension and preeclampsia — multiple trials have shown that L-arginine 3–4 g/day in pregnancy reduces the incidence and severity of preeclampsia, particularly in women with prior history. The mechanism is thought to involve restoration of placental endothelial NO production, which is impaired in preeclampsia.
- Salt-sensitive hypertension — salt-sensitive individuals tend to have impaired renal NO production, and arginine repletion may be especially helpful in this subset
- Hypertension with metabolic syndrome — the combination of hypertension, insulin resistance, dyslipidemia, and obesity is associated with elevated ADMA and reduced arginine bioavailability; supplementation has shown modest but reproducible benefit
- Hypertension in chronic kidney disease — complicated by elevated ADMA from impaired renal clearance; arginine supplementation requires nephrology coordination because of the risk of urea accumulation
The AHA does not endorse arginine as a stand-alone antihypertensive intervention, but it does acknowledge the cardiovascular literature in its scientific statements on functional foods and supplements. Most cardiologists who incorporate arginine into practice do so as a complementary intervention alongside conventional medication, lifestyle modification, and management of contributing conditions.
Peripheral Arterial Disease and Stable Angina
Peripheral arterial disease (PAD) is the clinical condition most consistently shown to benefit from arginine supplementation in randomized trials. PAD is characterized by atherosclerotic narrowing of the lower extremity arteries, producing intermittent claudication (calf, thigh, or buttock pain on walking that resolves with rest) and reduced ankle-brachial index. The underlying pathophysiology includes endothelial dysfunction, reduced NO bioavailability, and impaired functional hyperemia — precisely the mechanisms most directly addressed by arginine repletion.
The pivotal trial was by Boger and colleagues, published in Circulation in 1998. Forty PAD patients were randomized to oral L-arginine 16 g/day, oral prostaglandin E1 (the standard pharmacological intervention at the time), or placebo, for three weeks. The L-arginine group showed significant improvements in pain-free walking distance (approximately 230%), absolute walking distance (approximately 155%), and quality-of-life measures, with effect sizes comparable to or exceeding the prostaglandin arm.
Subsequent trials have confirmed the benefit, though typically at more practical doses (3–9 g/day) and with effect sizes proportional to dose. The meta-analysis by Jablecka et al. (2012) of 12 PAD trials found pooled improvement in pain-free walking distance of approximately 60–100 meters over placebo, again with effect sizes comparable to the dedicated PAD medication pentoxifylline.
For stable angina pectoris, the evidence is more mixed. Small trials have shown improvements in exercise tolerance, time to ST-segment depression, and quality of life with arginine supplementation in patients with chronic stable angina, but the effect sizes are smaller than in PAD, and not all trials have replicated the benefit. The TOTAL-AMI and similar trials in acute coronary syndrome have been mostly negative or showed harm signals (discussed below). Current cardiology practice does not include arginine in standard angina management protocols, but individual patients may use it adjunctively.
For more on cardiovascular disease, see our Cardiovascular Disease page and the related Hypertension page.
Heart Failure and the VINTAGE-MI Trial
The arginine story in heart failure is one of the clearest illustrations of why bench biology and clinical trials must both be examined before any supplement is broadly recommended. Animal and small human studies suggested that arginine could improve cardiac output, reduce systemic vascular resistance, and provide hemodynamic benefit in heart failure. Then the VINTAGE-MI trial (Schulman et al., JAMA 2006) tested L-arginine 3 g three times daily in 153 patients after acute myocardial infarction.
The results were striking and unexpected. The arginine group showed no improvement in left ventricular ejection fraction or vascular stiffness compared with placebo. More troublingly, there were six deaths in the arginine group versus zero in the placebo group over the six-month follow-up. The trial was stopped early by the data safety monitoring board. The deaths could not be definitively attributed to arginine (the small numbers and possible chance imbalance limit interpretation), but the safety signal was strong enough to bring routine post-MI arginine supplementation to a halt.
Several explanations have been proposed:
- Peroxynitrite generation — in the inflamed, oxidatively-stressed post-MI myocardium, NO produced from arginine may react with superoxide to form peroxynitrite, a highly cytotoxic oxidant that worsens myocardial injury
- NOS uncoupling — under conditions of BH4 depletion (which occurs in post-MI tissue), arginine-driven NOS activity may shunt toward superoxide production rather than NO, again worsening oxidative damage
- Hemodynamic vulnerability — aggressive vasodilation in patients with marginal cardiac output may compromise coronary perfusion pressure and worsen ischemia
The clinical takeaway: L-arginine should not be used in the acute post-MI period, and patients with significant heart failure should use arginine only under cardiology supervision. The L-arginine paradox in heart failure refers specifically to this disconnect — the substrate that should restore endothelial function actually causes harm in certain populations because of the broader oxidative biology.
Clinical Practice Recommendations
Based on the published evidence, a defensible practical approach to arginine for cardiovascular health:
- Endothelial dysfunction without overt disease — consider L-citrulline 3–6 g/day as a more bioavailable alternative to L-arginine. Reassess after 8–12 weeks with flow-mediated dilation if available, or with clinical markers (blood pressure, exercise tolerance).
- Mild to moderate hypertension — L-citrulline 3–6 g/day as adjunct to lifestyle modification and standard antihypertensive therapy. Expect 4–6 mmHg systolic reduction; this is meaningful but not a substitute for primary medication if BP is significantly elevated.
- Peripheral arterial disease with claudication — the strongest evidence base. L-arginine 6–9 g/day or L-citrulline 3–6 g/day, combined with supervised exercise therapy and standard cardiovascular risk-factor management. Expect meaningful improvement in pain-free walking distance over 8–12 weeks.
- Stable chronic angina — can be used adjunctively at 3–6 g/day arginine or 3 g/day citrulline, but evidence is weaker than for PAD. Discontinue if no benefit at 12 weeks.
- Gestational hypertension prevention — L-arginine 3 g/day starting in second trimester, in consultation with obstetrician. Multiple trials show reduced incidence of preeclampsia.
- Post-MI / acute coronary syndrome — avoid. Based on the VINTAGE-MI safety signal, arginine should not be used in the first 6 months after acute MI.
- Heart failure (NYHA III-IV) — use only under cardiology supervision. The evidence is inconsistent and there is a plausible mechanism for harm.
- Active herpes simplex infection — arginine is required for HSV replication and can precipitate outbreaks. Patients with frequent herpes recurrences should use citrulline preferentially and may benefit from concurrent lysine supplementation; see L-Lysine.
Cautions and Contraindications
- Recent myocardial infarction (within 6 months) — absolute contraindication based on VINTAGE-MI mortality signal. Consult cardiology before any use.
- Concurrent nitrate medication — nitroglycerin, isosorbide, and other nitrates donate NO directly; combined use with arginine can produce excessive vasodilation, hypotension, and reflex tachycardia. Use only with prescriber awareness.
- Concurrent phosphodiesterase-5 inhibitors — sildenafil, tadalafil, vardenafil prolong the cGMP signal downstream of NO; combined use with arginine can amplify hypotensive effects. Generally safe at standard doses but caution in patients with marginal blood pressure.
- Active herpes simplex infection — arginine is required for HSV replication and can precipitate outbreaks. Discussed above.
- Chronic kidney disease stages 3–5 — impaired clearance of arginine metabolites, particularly urea and creatinine; coordinate with nephrology.
- Asthma — theoretical concern that arginine could feed pathological iNOS activity in airway inflammation, but small studies have actually shown benefit from arginine in asthma. Discuss with pulmonologist.
- Gastrointestinal tolerance — oral arginine doses above 5–10 g often cause nausea, cramping, and diarrhea. Citrulline is better tolerated at equivalent functional doses.
Key Research Papers
- Boger RH et al. (1998). Restoring vascular nitric oxide formation by L-arginine improves the symptoms of intermittent claudication in patients with peripheral arterial occlusive disease. Circulation. — PubMed
- Schulman SP et al. (2006). L-arginine therapy in acute myocardial infarction: the Vascular Interaction With Age in Myocardial Infarction (VINTAGE MI) randomized clinical trial. JAMA. — PubMed
- Dong JY et al. (2011). Effect of oral L-arginine supplementation on blood pressure: a meta-analysis of randomized, double-blind, placebo-controlled trials. American Heart Journal. — PubMed
- Schwedhelm E et al. (2008). Pharmacokinetic and pharmacodynamic properties of oral L-citrulline and L-arginine. British Journal of Clinical Pharmacology. — PubMed
- Moinard C et al. (2008). Dose-ranging effects of citrulline administration on plasma amino acids and hormonal patterns in healthy subjects. British Journal of Nutrition. — PubMed
- Boger RH (2007). The pharmacodynamics of L-arginine. Journal of Nutrition. — PubMed
- Vallance P, Leiper J (2004). Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arteriosclerosis, Thrombosis, and Vascular Biology. — PubMed
- Cooke JP (2003). NO and angiogenesis. Atherosclerosis Supplements. — PubMed
- Forstermann U, Sessa WC (2012). Nitric oxide synthases: regulation and function. European Heart Journal. — PubMed
- Hayashi T et al. (2005). L-Citrulline and L-arginine supplementation retards the progression of high-cholesterol-diet-induced atherosclerosis in rabbits. PNAS. — PubMed
- Bode-Boger SM et al. (2003). The L-arginine paradox: importance of the L-arginine/asymmetrical dimethylarginine ratio. Pharmacology and Therapeutics. — PubMed
- Jablecka A et al. (2012). The effect of oral L-arginine supplementation on fasting glucose, HbA1c, nitric oxide and total antioxidant status in diabetic patients with atherosclerotic peripheral arterial disease. European Review for Medical and Pharmacological Sciences. — PubMed
- Morris SM Jr (2009). Recent advances in arginine metabolism: roles and regulation of the arginases. British Journal of Pharmacology. — PubMed
- Loscalzo J (2004). L-Arginine and atherothrombosis. Journal of Nutrition. — PubMed
PubMed Topic Searches
- PubMed: L-arginine and endothelial NO
- PubMed: ADMA and cardiovascular risk
- PubMed: Citrulline supplementation
- PubMed: Arginine for PAD/claudication
- PubMed: The arginine paradox
- PubMed: Arginase in cardiovascular disease
Connections
- Arginine Overview
- Arginine Benefits Hub
- Arginine for Erectile Function
- Arginine for Wound Healing
- Arginine for Immune Function
- Cardiovascular Disease
- Hypertension
- Blood Pressure
- Lysine
- Lysine
- Creatine
- Kidney Function
- Metabolic Syndrome
- Diabetes
- Taurine
- All Amino Acids
- Citrulline — the more bioavailable nitric-oxide precursor compared throughout this page.