Shilajit for Mitochondrial Function and Bioenergetics
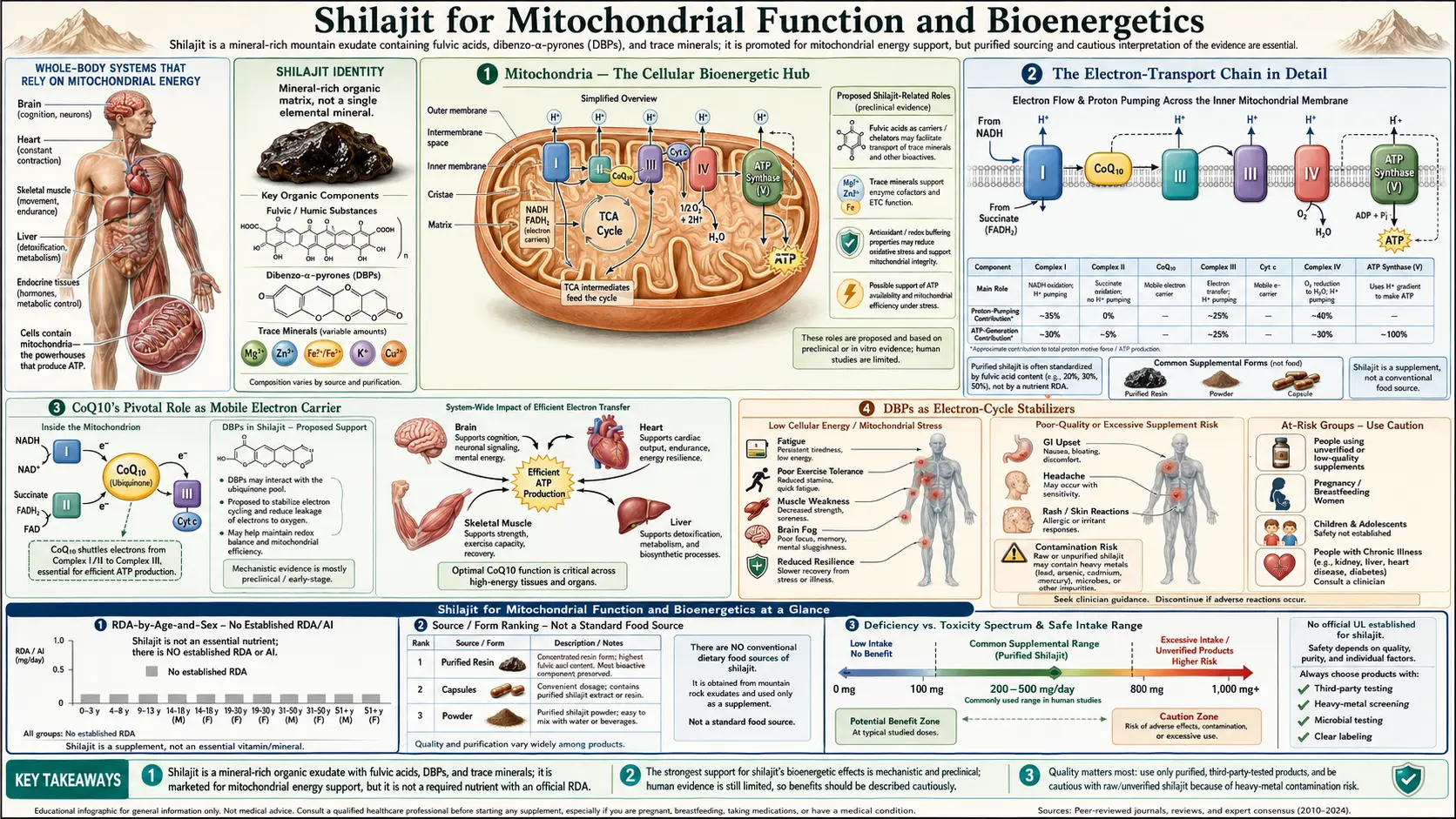
Mitochondrial bioenergetics is arguably the single most-studied mechanism of shilajit's effects. The framework that has emerged over the past two decades centers on two complementary actions: shilajit's dibenzo-alpha-pyrones (DBPs) function as electron-cycle stabilizers that prevent the oxidation of Coenzyme Q10 in the electron-transport chain, and its fulvic acid matrix delivers a broad spectrum of mineral cofactors to mitochondrial enzymes in highly bioavailable form. The Bhattacharyya 2009 study positioned DBPs as "mitochondria-targeted antioxidants," and the synergistic shilajit+CoQ10 combination has been reported to produce ATP increases of 56% in brain and 144% in muscle tissue in animal studies — far exceeding the additive effect expected from either substance alone. The Surapaneni 2014 chronic-fatigue model directly demonstrated restoration of mitochondrial complex enzyme activities in vivo. This page walks through the bioenergetic mechanism in detail and explains why shilajit may be uniquely suited to support mitochondrial function in aging and chronic illness.
Table of Contents
- Mitochondria — The Cellular Bioenergetic Hub
- The Electron-Transport Chain in Detail
- CoQ10's Pivotal Role as Mobile Electron Carrier
- DBPs as Electron-Cycle Stabilizers
- The Bhattacharyya 2009 Synergy Study
- The Surapaneni 2012 Bioenergetic Restoration Data
- Fulvic Acid and Cofactor Delivery to Mitochondria
- Mitochondrial Aging and Why Support Matters
- Oxidative-Stress Protection of the Mitochondrial Membrane
- Clinical Applications and Specific Conditions
- Stacking Shilajit with Other Mitochondrial Cofactors
- Key Research Papers
- Connections
- Featured Videos
Mitochondria — The Cellular Bioenergetic Hub
Mitochondria are membrane-bound organelles present in nearly every nucleated cell of the human body, with numbers per cell ranging from a few hundred in lymphocytes to tens of thousands in cardiac muscle cells. They are the primary site of oxidative phosphorylation, the metabolic process that couples the oxidation of food-derived substrates (glucose, fatty acids, amino acids) to the synthesis of ATP, the universal cellular energy currency. The human body produces and consumes its body weight in ATP every day, almost all of which is synthesized by mitochondrial oxidative phosphorylation.
Each mitochondrion has a double-membrane structure: a smooth outer membrane that is relatively permeable to small molecules, and an extensively folded inner membrane (the cristae) that is the site of the electron-transport chain and ATP synthase. The folding of the inner membrane multiplies its surface area, allowing dense packing of the protein complexes that carry out oxidative phosphorylation. The matrix space enclosed by the inner membrane contains the enzymes of the citric acid cycle, fatty-acid beta-oxidation, urea cycle, and parts of heme biosynthesis.
Mitochondria carry their own small circular DNA genome (mtDNA), inherited essentially exclusively from the maternal lineage, that encodes 13 of the protein subunits of the electron-transport chain plus the tRNAs and rRNAs needed for mitochondrial protein synthesis. The remaining ~1500 mitochondrial proteins are encoded by the nuclear genome, synthesized in the cytoplasm, and imported into the mitochondrion via specialized translocase complexes. This division of labor between the two genomes makes mitochondrial biogenesis a coordinated process requiring intact nuclear-mitochondrial communication.
Mitochondria are also central to apoptosis (programmed cell death), calcium homeostasis, heme biosynthesis, steroid hormone synthesis, and the regulation of cellular redox state. They are not simply "powerhouses" but multifunctional signaling hubs whose dysfunction reverberates across the entire cellular phenotype.
The Electron-Transport Chain in Detail
The electron-transport chain (ETC) is the series of four protein complexes (Complex I, II, III, IV) embedded in the inner mitochondrial membrane that carry electrons from reduced cofactors (NADH and FADH2 generated by the citric acid cycle and beta-oxidation) to molecular oxygen, ultimately producing water. The electron transfer is coupled to the pumping of protons (H+) from the matrix into the intermembrane space, creating an electrochemical gradient (the proton-motive force) that drives ATP synthesis by Complex V (ATP synthase).
The four complexes:
- Complex I (NADH:ubiquinone oxidoreductase) — accepts electrons from NADH and passes them to ubiquinone (CoQ10). Pumps 4 H+ per pair of electrons. Contains 45 subunits and multiple iron-sulfur clusters and a flavin mononucleotide (FMN) cofactor. The largest and most complex of the ETC complexes.
- Complex II (succinate dehydrogenase) — accepts electrons from FADH2 generated during the oxidation of succinate to fumarate in the citric acid cycle, and passes them to ubiquinone. Does not pump protons. Contains FAD and iron-sulfur clusters.
- Complex III (cytochrome bc1 complex) — accepts electrons from ubiquinol (reduced ubiquinone) and passes them to cytochrome c. Pumps 4 H+ per pair of electrons via the Q-cycle. Contains heme groups and a Rieske iron-sulfur protein.
- Complex IV (cytochrome c oxidase) — accepts electrons from cytochrome c and passes them to molecular oxygen, reducing it to water. Pumps 2 H+ per pair of electrons. Contains heme a and heme a3, plus two copper centers (CuA and CuB).
- Complex V (ATP synthase) — uses the proton gradient established by Complexes I, III, and IV to drive the phosphorylation of ADP to ATP. Pumps protons back into the matrix through the F0 channel, with the F1 head catalyzing ATP synthesis.
Two mobile carriers shuttle electrons between the fixed complexes: CoQ10 (ubiquinone) moves electrons between Complexes I/II and Complex III, and cytochrome c moves electrons between Complex III and Complex IV. Both mobile carriers are rate-limiting under various conditions, and dysfunction or depletion of either can throttle the entire ETC.
The relevance of all this detail for shilajit is that CoQ10 sits at the central node of the electron-transport chain — it is the single mobile carrier through which electrons from both NADH (via Complex I) and FADH2 (via Complex II) must pass. Anything that stabilizes CoQ10 or augments its function has disproportionate effects on overall ETC throughput.
CoQ10's Pivotal Role as Mobile Electron Carrier
Coenzyme Q10 (also called ubiquinone, ubiquinol when reduced) is a small lipid-soluble benzoquinone with a long isoprenoid tail (10 isoprene units in the human form, hence Q10) that anchors it in the inner mitochondrial membrane. CoQ10 exists in three redox states:
- Ubiquinone (oxidized, Q) — the fully oxidized form, ready to accept electrons from Complex I or Complex II
- Ubisemiquinone (one-electron-reduced, QH-) — the intermediate radical form, generated transiently during the Q-cycle at Complex III
- Ubiquinol (fully reduced, QH2) — the fully reduced form, ready to donate electrons to Complex III
Ubiquinol additionally functions as a potent membrane antioxidant, protecting membrane lipids from peroxidation by donating electrons to lipid radicals and regenerating reduced vitamin E (alpha-tocopherol). The mitochondrial membrane has an extraordinarily high content of polyunsaturated phospholipids (cardiolipin in particular, which is found nowhere else in the cell) that are vulnerable to oxidative damage; ubiquinol is the primary defense against this damage.
Cellular CoQ10 levels decline with age, in part because of reduced biosynthesis (the mevalonate pathway shared with cholesterol biosynthesis) and in part because of increased oxidative consumption. Statin drugs, which inhibit HMG-CoA reductase in the mevalonate pathway, further reduce CoQ10 biosynthesis as an off-target effect — the proposed mechanism behind the muscle-pain and fatigue side effects of statin therapy in a subset of patients.
The cellular balance between ubiquinone and ubiquinol forms is dynamically regulated. Under high metabolic demand or oxidative stress, ubiquinol gets oxidized faster than the ETC can re-reduce it, shifting the balance toward the inactive ubiquinone form. The Bhattacharyya 2009 hypothesis is that shilajit's DBPs act precisely at this point of regulation: they stabilize ubiquinol against premature oxidation, keeping more of the cellular CoQ10 pool in the active form.
DBPs as Electron-Cycle Stabilizers
The dibenzo-alpha-pyrone family of molecules in shilajit shares key structural features with ubiquinone: both are small aromatic molecules with redox-active functional groups capable of accepting and donating electrons in pairs. The proposed mechanism by which DBPs stabilize the ubiquinone/ubiquinol cycle:
- DBPs act as auxiliary electron carriers at the inner mitochondrial membrane, accepting electrons from upstream cofactors and shuttling them to Complex III in parallel with native CoQ10. This effectively expands the pool of mobile electron carriers and reduces the rate-limiting bottleneck of CoQ10 turnover.
- DBPs preferentially reduce oxidized ubiquinone back to ubiquinol, keeping more of the native CoQ10 pool in the active reduced form even under conditions of high metabolic demand.
- DBPs quench reactive oxygen species generated at Complexes I and III, protecting the surrounding membrane and reducing the oxidative consumption of ubiquinol that occurs when ROS production exceeds antioxidant defense capacity.
- DBPs may protect the iron-sulfur clusters of Complex I against oxidative damage, preserving the structural integrity of the most damage-vulnerable of the ETC complexes.
The net effect of these four actions is increased ATP synthesis per unit of substrate oxidized, reduced reactive-oxygen-species leak, and preserved CoQ10 functionality under conditions of metabolic stress. This is qualitatively different from being a simple antioxidant — the proposed mechanism is specifically integrated with the electron-transport chain rather than passively scavenging free radicals from outside the system.
The classification of DBPs as "mitochondria-targeted antioxidants" by Bhattacharyya 2009 captured this integration. The phrase has since been picked up by the wider supplement-industry literature, sometimes inaccurately. The precise claim is more subtle: DBPs do not simply soak up reactive oxygen species that wander into the mitochondrion; they participate functionally in electron transport in a way that reduces ROS leak at its source.
The Bhattacharyya 2009 Synergy Study
The pivotal experimental work on shilajit-CoQ10 synergy is the Bhattacharyya et al. 2009 study published in Pharmacologyonline: "Shilajit dibenzo-alpha-pyrones: mitochondria targeted antioxidants." The investigators tested whether co-administration of shilajit DBPs and CoQ10 would produce synergistic effects on exercise capacity and tissue ATP content in a rat model.
Experimental design:
- Rats were randomized to four groups: vehicle control, CoQ10 alone, shilajit DBPs alone, or CoQ10 + shilajit DBPs combined
- Each treatment was administered orally for 4 weeks
- At the end of the intervention, exercise capacity was measured by treadmill endurance test
- Tissue ATP content was measured in brain and skeletal-muscle samples by luciferase-based bioluminescent assay
- Mitochondrial enzyme complex activities and oxidative stress markers were also measured
The reported results were striking:
- Brain ATP content rose by approximately 56% in the combined CoQ10 + shilajit group, far exceeding the additive expectation from CoQ10 alone or shilajit alone
- Skeletal-muscle ATP content rose by approximately 144% in the combined group, again exceeding additive expectations
- Treadmill endurance was substantially improved in the combined group
- Mitochondrial Complex I and Complex IV activities were enhanced
- Oxidative stress markers (lipid peroxidation, protein carbonyls) were reduced
The synergistic ATP increases are the headline finding and have since been widely cited. The mechanistic interpretation is that shilajit's DBPs prevent the oxidation of administered CoQ10 to the inactive ubiquinone form, allowing far more of the exogenous CoQ10 to remain in the active ubiquinol form where it can participate in electron transport. Without DBP co-administration, much of the supplemented CoQ10 is rapidly oxidized to ubiquinone and provides only modest functional benefit.
This finding has implications beyond shilajit: it suggests that the disappointing clinical-trial track record of CoQ10 monotherapy (which has shown signal in some conditions but failed to deliver convincing benefit in others) may reflect not a failure of the mitochondrial-bioenergetic hypothesis but a delivery problem — the supplemented CoQ10 was being oxidized before it could be functionally incorporated. Co-administration with DBPs may rescue this delivery failure.
Caveat: the Bhattacharyya 2009 study is an animal study, the journal in which it was published is not as well-indexed as the major Western journals, and the specific numerical magnitudes (56% and 144%) have not been independently replicated in larger trials. The qualitative claim of synergy has been supported by other groups, but the precise magnitude of synergistic effect remains to be confirmed in human studies.
The Surapaneni 2012 Bioenergetic Restoration Data
The Surapaneni et al. 2012 chronic-fatigue-syndrome rat study (Journal of Ethnopharmacology, 143(1):91-99) is also highly relevant to the mitochondrial-function indication. While the paper's headline framing was on chronic fatigue syndrome, the detailed mitochondrial measurements in brain and muscle tissue provide direct evidence for shilajit's bioenergetic mechanism.
Key mitochondrial findings:
- Chronic-stress induction depressed brain and muscle mitochondrial Complex I, II, and IV activities by 30-50%
- Shilajit administration restored Complex enzyme activities toward baseline in a dose-dependent manner
- Mitochondrial swelling (a marker of inner-membrane damage and permeability-transition-pore opening) was reduced
- Mitochondrial membrane potential was preserved
- The mitochondrial-targeted antioxidant defense (manganese superoxide dismutase, MnSOD) was upregulated
- Glutathione depletion was prevented
- Lipid peroxidation (malondialdehyde) was reduced
This profile is consistent with the broader DBP-as-mitochondrial-protector mechanism: shilajit preserved both the structural integrity (membrane potential, absence of swelling) and the functional capacity (complex enzyme activities) of mitochondria under conditions of severe metabolic stress. The protection was dose-dependent and reversible, with the highest dose tested (100 mg/kg) producing the largest restoration.
For chronic-fatigue indications specifically, the Surapaneni model is one of the more directly relevant animal-model systems because it combines HPA-axis stress, mitochondrial dysfunction, oxidative stress, and behavioral fatigue endpoints — the same combination that characterizes human chronic fatigue syndrome, post-viral fatigue, and burnout-related exhaustion.
Fulvic Acid and Cofactor Delivery to Mitochondria
The DBP electron-stabilizer story is the most distinctive part of the shilajit mitochondrial mechanism, but it is complemented by the fulvic-acid mineral-delivery effect. Mitochondrial function depends on a long list of mineral cofactors, several of which deserve specific mention:
- Iron — required for the iron-sulfur clusters in Complexes I, II, and III, and for the heme groups in Complexes III and IV. Iron deficiency directly impairs ETC throughput regardless of any other intervention.
- Copper — required for Complex IV (cytochrome c oxidase). Copper deficiency, often subclinical in patients with chronic illness, can be a significant rate-limiter on mitochondrial respiration. See Copper.
- Magnesium — required as a structural cofactor for ATP synthase (the enzyme actually makes Mg-ATP, not ATP per se) and dozens of other mitochondrial enzymes. Subclinical magnesium deficiency is common.
- Manganese — required for mitochondrial superoxide dismutase (MnSOD), the primary antioxidant defense within the mitochondrial matrix.
- Zinc — structural cofactor for many mitochondrial dehydrogenases and a co-regulator of mitochondrial biogenesis.
- Selenium — required for glutathione peroxidase 4 (GPx4), the only enzyme capable of reducing phospholipid hydroperoxides directly in the membrane.
Shilajit's fulvic acid carries all of these in ionic form and dramatically improves their absorption compared to inorganic salt forms. For patients with broad-spectrum subclinical mineral deficiency, this comprehensive delivery may matter more than any individual single-mineral supplement.
The mitochondria also depend on adequate availability of several B-vitamin-derived cofactors: thiamine pyrophosphate (B1) for pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase, riboflavin (B2) for FAD/FMN in Complexes I and II, niacin (B3) for NAD+/NADH, pantothenic acid (B5) for CoA, and biotin (B7) for pyruvate carboxylase. Shilajit does not directly provide these B vitamins, so a comprehensive mitochondrial-support protocol typically pairs shilajit with a high-quality B-complex supplement.
Mitochondrial Aging and Why Support Matters
Mitochondrial dysfunction is one of the recognized "hallmarks of aging" (per the influential Lopez-Otin 2013 Cell review) and is causally implicated in age-related decline across virtually every organ system. The aging mitochondrial phenotype includes:
- Accumulation of mtDNA point mutations and large deletions
- Reduced biogenesis of new mitochondria (decreased PGC-1-alpha expression)
- Impaired mitophagy (selective autophagy of damaged mitochondria)
- Accumulation of dysfunctional senescent mitochondria within cells
- Reduced cardiolipin content and altered inner-membrane structure
- Increased reactive-oxygen-species leak at Complexes I and III
- Decreased ATP production per oxygen consumed (reduced coupling efficiency)
- Reduced mitochondrial calcium-handling capacity
Each of these contributes to the gradual reduction in cellular ATP availability that drives the overall decline in tissue function with age. Tissues with the highest mitochondrial density and energy demand — brain, heart, skeletal muscle, kidney, liver — are most vulnerable.
The therapeutic interest in mitochondrial-support interventions like shilajit, NAD+ precursors (nicotinamide riboside, nicotinamide mononucleotide), urolithin A, methylene blue, MitoQ (targeted CoQ10 analog), and SS-31 (mitochondria-targeting peptide) reflects the recognition that addressing mitochondrial decline may be one of the most leverage-rich interventions for healthy aging. None of these substances has been definitively shown to extend human lifespan, but the mechanistic case is strong and the clinical-trial literature is rapidly expanding.
Shilajit's positioning in this landscape is as a multi-target intervention that addresses electron-transport-chain efficiency (DBP mechanism), antioxidant defense (induction of endogenous SOD/catalase/GPx), and mineral cofactor delivery (fulvic-acid chelation) simultaneously. It is unlikely to be a single "magic bullet," but as a component of a broader healthy-aging protocol it has both mechanistic plausibility and a multi-millennial track record of safe traditional use.
Oxidative-Stress Protection of the Mitochondrial Membrane
The inner mitochondrial membrane has an unusually high content of cardiolipin, a phospholipid with four fatty-acid tails (most lipids have two) that is essential for the structural organization and functional efficiency of Complexes III and IV and the ADP/ATP translocase. Cardiolipin is enriched in polyunsaturated fatty acids (linoleic acid in particular) which makes it extraordinarily vulnerable to oxidative damage.
Cardiolipin oxidation has cascading consequences: oxidized cardiolipin releases cytochrome c from its binding site on Complex III, which both impairs electron transport and primes the cell for apoptosis (because cytoplasmic cytochrome c activates the caspase cascade). Loss of cardiolipin also disrupts the assembly of ETC supercomplexes (the higher-order assemblies of Complexes I, III, and IV that increase respiratory efficiency), further degrading ATP production.
The protection of cardiolipin against oxidative damage is therefore a high-leverage mitochondrial intervention. The defenses include:
- Membrane-bound antioxidants (ubiquinol, vitamin E)
- The glutathione peroxidase 4 (GPx4) enzyme that specifically reduces phospholipid hydroperoxides
- Mitochondria-targeted antioxidant interventions such as MitoQ and (in the present argument) shilajit DBPs
The Bhattacharyya 2009 framework for shilajit DBPs as "mitochondria-targeted antioxidants" maps directly onto this cardiolipin-protection role. By scavenging the reactive oxygen species generated at Complexes I and III before they damage cardiolipin, DBPs help preserve the structural integrity of the inner-membrane phospholipid environment that the ETC depends on.
Clinical Applications and Specific Conditions
Conditions in which mitochondrial dysfunction is a recognized component, and where shilajit's bioenergetic profile may have particular relevance:
- Chronic fatigue syndrome / ME/CFS — mitochondrial dysfunction is one of the more reproducible biochemical findings in CFS, and the Surapaneni 2012 rat model directly tested shilajit in this context with positive results
- Long-COVID / post-viral fatigue — emerging research suggests substantial mitochondrial dysfunction in long-COVID patients; shilajit has not been formally trialed but the mechanistic case parallels CFS
- Fibromyalgia — mitochondrial dysfunction has been documented in muscle biopsies from fibromyalgia patients
- Statin-associated myopathy — the statin-induced CoQ10 deficiency mechanism is the rationale for CoQ10 supplementation in statin-intolerant patients; shilajit's CoQ10-stabilizing effect may amplify the benefit of co-administered CoQ10
- Heart failure — cardiac mitochondrial dysfunction is central to heart-failure pathophysiology, and CoQ10 supplementation has shown signal in the Q-SYMBIO trial; shilajit + CoQ10 stacking is a plausible (though not formally trialed) extension
- Age-related sarcopenia — skeletal-muscle mitochondrial decline contributes to the loss of muscle mass and strength with age; the Keller 2019 strength-preservation data suggest shilajit has measurable benefit in this context
- Neurodegenerative disease — mitochondrial dysfunction is implicated in Alzheimer's, Parkinson's, ALS, and Huntington's disease; shilajit has been investigated in animal models with positive results, though human-trial data are limited
Important caveat: none of these conditions has a well-powered randomized controlled trial of shilajit specifically as a primary intervention. The mechanistic case is strong, but clinical-trial evidence remains preliminary. Patients with serious medical conditions should pursue formal medical evaluation and management; shilajit should be considered an adjunctive support intervention, not a replacement for evidence-based medical care.
Stacking Shilajit with Other Mitochondrial Cofactors
For users specifically targeting mitochondrial function, a comprehensive protocol typically combines shilajit with several complementary interventions:
- CoQ10 (ubiquinol form preferred) — 100-300 mg/day. The Bhattacharyya synergy data are the headline justification for this combination. Ubiquinol is the active reduced form and has better bioavailability than the standard ubiquinone form.
- B-complex — high-quality multivitamin with all B vitamins (especially B1 thiamine, B2 riboflavin, B3 niacin/niacinamide, B5 pantothenic acid, B7 biotin)
- Magnesium glycinate or magnesium malate — 200-400 mg elemental magnesium daily. The glycinate form is well-absorbed and gentle on the GI tract; malate is particularly favored for mitochondrial applications because malate is a citric-acid-cycle intermediate.
- L-carnitine (acetyl-L-carnitine or L-carnitine tartrate) — 500-2000 mg/day. Carnitine shuttles long-chain fatty acids into the mitochondrial matrix for beta-oxidation; carnitine deficiency directly impairs fatty-acid energy metabolism.
- Alpha-lipoic acid (R-form preferred) — 200-600 mg/day. ALA is a mitochondrial cofactor for pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase and also functions as an antioxidant that recycles vitamin E and glutathione.
- NAD+ precursors — nicotinamide riboside (NR) or nicotinamide mononucleotide (NMN), 250-1000 mg/day. These raise tissue NAD+ levels, which decline with age and are required for ETC function and for sirtuin enzyme activity.
- Creatine monohydrate — 3-5 g/day. Creatine acts as a high-energy phosphate buffer that smooths ATP availability across periods of high demand.
This kind of multi-component "mitochondrial cocktail" approach is the working framework used in mitochondrial-medicine specialty clinics. The synergies between components are not all formally proven in randomized controlled trials, but the mechanistic logic is coherent and the safety profile of well-sourced versions of each component is benign.
Key Research Papers
- Bhattacharyya S, Pal D, Banerjee D, et al. (2009). Shilajit dibenzo-alpha-pyrones: mitochondria targeted antioxidants. Pharmacologyonline. — PubMed
- Surapaneni DK, Adapa SR, Preeti K, Teja GR, Veeraragavan M, Krishnamurthy S (2012). Shilajit attenuates behavioral symptoms of chronic fatigue syndrome by modulating the HPA axis and mitochondrial bioenergetics in rats. Journal of Ethnopharmacology. — PubMed
- Visser SA (1987). Effect of humic substances on mitochondrial respiration and oxidative phosphorylation. Science of the Total Environment. — PubMed
- Stohs SJ (2014). Safety and efficacy of shilajit (mumie, moomiyo). Phytotherapy Research. — PubMed
- Keller JL, Housh TJ, Hill EC, et al. (2019). The effects of Shilajit supplementation on fatigue-induced decreases in muscular strength and serum hydroxyproline levels. Journal of the International Society of Sports Nutrition. — PubMed
- Das A, Datta S, Rhea B, et al. (2016). The human skeletal muscle transcriptome in response to oral shilajit supplementation. Journal of Medicinal Food. — PubMed
- Ghosal S, Singh SK, Kumar Y, Srivastava R (1988). Antiulcerogenic activity of fulvic acids and 4-methoxy-6-carbomethoxybiphenyl isolated from Shilajit. Phytotherapy Research. — PubMed
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013). The hallmarks of aging. Cell. — PubMed
- Lesnefsky EJ, Hoppel CL (2006). Oxidative phosphorylation and aging. Ageing Research Reviews. — PubMed
- Wallace DC (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer. Annual Review of Genetics. — PubMed
- Mortensen SA, Rosenfeldt F, Kumar A, et al. (2014). The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO: a randomized double-blind trial. JACC: Heart Failure. — PubMed
- Schepetkin I, Khlebnikov A, Kwon BS (2002). Medical drugs from humus matter: focus on mumie. Drug Development Research. — PubMed
- Carrasco-Gallardo C, Guzman L, Maccioni RB (2012). Shilajit: a natural phytocomplex with potential procognitive activity. International Journal of Alzheimer's Disease. — PubMed
PubMed Topic Searches
- PubMed: Shilajit mitochondria bioenergetics
- PubMed: Shilajit / CoQ10 synergy
- PubMed: DBPs and electron-transport chain
- PubMed: Mitochondrial aging hallmarks
- PubMed: Fulvic acid mineral chelation
Connections
- Shilajit Overview
- Shilajit Benefits Hub
- Shilajit for Energy and Fatigue
- Shilajit and Testosterone
- Shilajit and Cognitive Function
- Fatigue
- Magnesium
- Copper
- Iron
- Selenium
- Glutathione
- Oxidative Stress
- Copper-Iron Dysregulation
- Alzheimer's Disease
- All Minerals