Hypersensitivity Pneumonitis
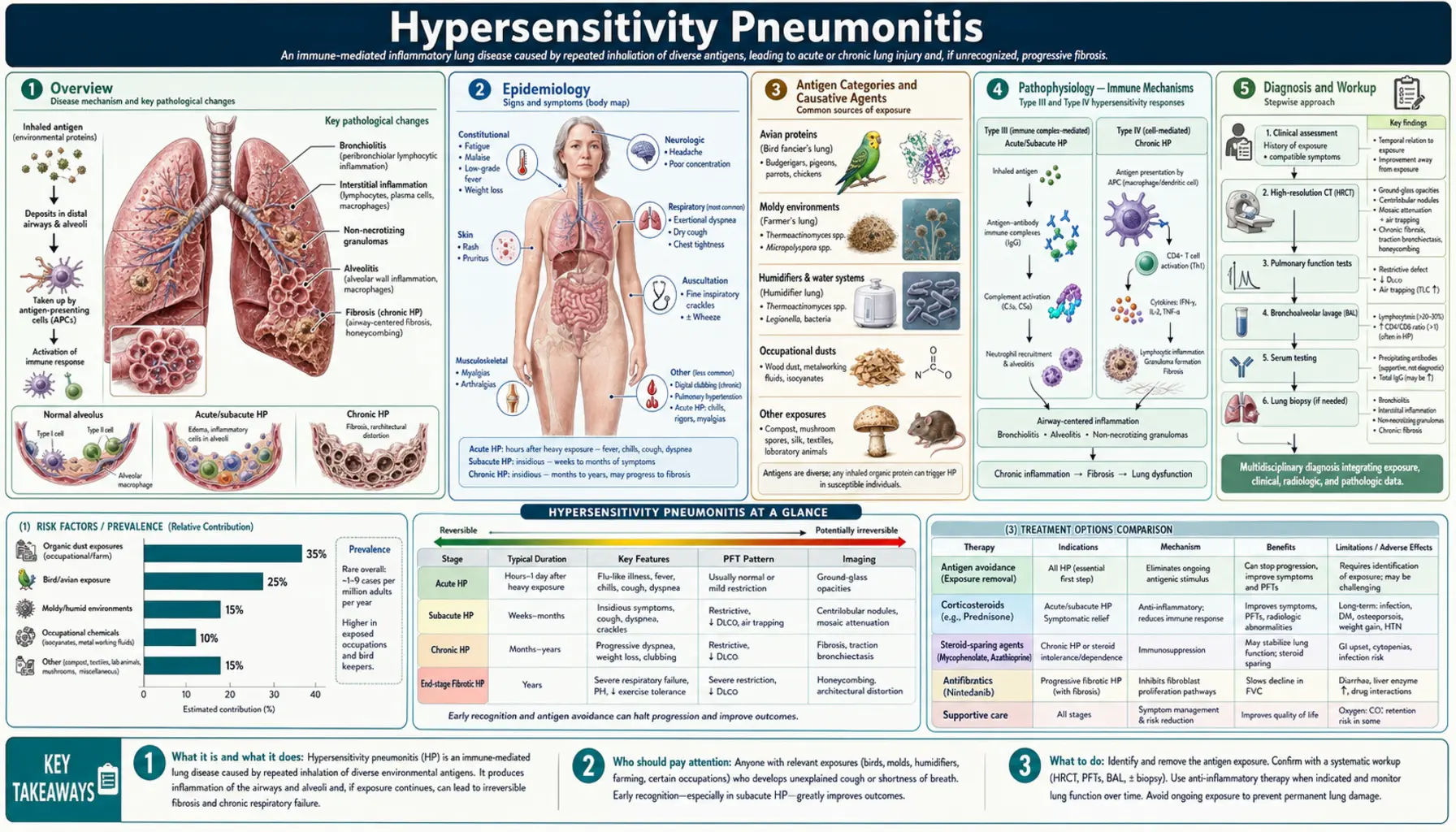
Table of Contents
- Overview
- Epidemiology
- Antigen Categories and Causative Agents
- Pathophysiology — Immune Mechanisms
- Clinical Phases — Acute, Subacute, Chronic
- Diagnosis — Imaging, BAL, and Biopsy
- Treatment — Antigen Avoidance and Corticosteroids
- Prognosis
- Prevention
- Research Papers
- Connections
- Featured Videos
Overview
Hypersensitivity pneumonitis (HP), also called extrinsic allergic alveolitis (EAA), is an immune-mediated interstitial lung disease caused by repeated inhalation of antigenic organic dusts or chemical haptens in susceptible individuals. The lungs mount an exaggerated immunological response to inhaled particles — not because the particles are inherently toxic, but because the immune system mistakenly treats them as threats requiring an aggressive inflammatory counterattack.
Unlike simple allergic reactions confined to the airways, HP involves both immune complex-mediated (type III hypersensitivity) and T-cell-mediated delayed hypersensitivity (type IV) mechanisms, affecting the alveoli and interstitium rather than just the bronchi. This dual immune involvement explains why HP can smolder for months or years before becoming clinically apparent, and why it can progress to irreversible pulmonary fibrosis when antigen exposure continues unchecked.
The disease is recognized in dozens of occupational and environmental settings — from farmers inhaling mold spores in hay, to pigeon breeders breathing in bird protein from droppings, to workers in metalworking plants exposed to contaminated coolants, to homeowners with mycobacterium-colonized indoor hot tubs. The name of the condition often reflects the exposure: Farmer's Lung, Bird Fancier's Lung, Hot Tub Lung, Cheese Worker's Lung, Humidifier Lung.
The two pillars of management are antigen identification and avoidance — the most critical intervention — and immunosuppression with corticosteroids for moderate-to-severe active disease. In the chronic fibrotic phase, antifibrotic agents such as nintedanib are emerging as additional tools. Early diagnosis and antigen removal before fibrosis develops is the key to preventing irreversible lung damage.
Epidemiology
The true prevalence of HP is difficult to establish because the disease is frequently misdiagnosed as recurrent pneumonia, asthma, or idiopathic pulmonary fibrosis, and because subclinical sensitization without clinical disease is far more common than overt HP. Estimates suggest HP accounts for approximately 4–15% of all interstitial lung disease diagnoses in referral centers.
Specific forms vary widely in prevalence by geography and occupation:
- Farmer's Lung: Affects 0.5–3% of exposed farmers in temperate climates where hay is stored in humid conditions. Most prevalent in Ireland, Finland, Scotland, and the American Midwest. Incidence has declined with better grain drying technology.
- Bird Fancier's Lung (Pigeon Breeder's Disease): The most common form of HP in many countries. Accounts for 60–70% of HP diagnoses in some European series. Affects pigeon racers, parakeet and parrot owners, and those who keep ornamental birds indoors.
- Hot Tub Lung: An increasingly recognized form in the United States, particularly affecting younger adults and children with access to indoor heated pools or spas colonized with Mycobacterium avium complex (MAC).
HP affects all ages including children and adolescents. Some series report a female predominance, likely reflecting exposure patterns (bird keeping as a hobby) rather than inherent biological susceptibility. Critically, only 5–15% of exposed, sensitized individuals develop clinical HP, highlighting that host genetic factors are essential cofactors. HLA-DPB1 alleles (particularly HLA-DPB1*0201) have been associated with increased susceptibility to Bird Fancier's Lung, while HLA-DR3 and HLA-B8 correlate with Farmer's Lung risk.
Smoking is an important epidemiological confounder: current smokers have a paradoxically lower acute HP incidence (nicotine suppresses macrophage activation and reduces precipitin responses), yet smokers who do develop HP have worse fibrotic outcomes. This "protective effect" of smoking dissolves entirely in the chronic phase.
Antigen Categories and Causative Agents
HP can be caused by hundreds of antigens across several broad categories. Identifying the specific causative antigen is clinically essential for antigen avoidance counseling.
Microbial Antigens — Bacteria and Fungi
- Thermophilic actinomycetes — Saccharopolyspora rectivirgula (formerly Micropolyspora faeni) and Thermoactinomyces vulgaris from moldy hay, grain, sugar cane, and mushroom compost. The classic cause of Farmer's Lung.
- Aspergillus species — A. fumigatus, A. clavatus — Malt Worker's Lung, Grain Handler's Lung.
- Mycobacterium avium complex (MAC) — Colonizes indoor heated pools and hot tubs when water is not adequately treated. MAC aerosols during use produce Hot Tub Lung, characterized by prominent mosaic attenuation on CT and a younger patient demographic.
- Penicillium species — P. casei (Cheese Worker's Lung), P. frequentans (Suberosis — cork dust disease), humidifier lung.
- Aureobasidium pullulans and other fungi — Sauna Taker's Disease, contaminated humidifiers and air handling units.
Avian Proteins
- Bird droppings, feathers, and serum proteins — Pigeon Breeder's Lung (most common), Parakeet/Budgerigar Fancier's Lung, Parrot Lung, Turkey Handler's Disease. The antigens are avian IgA and IgG (serum proteins), mucin-1 antigen, and bloom (a waxy powder coating feathers). Bird Fancier's Lung is the most common cause of chronic HP leading to fibrosis in Europe and is the most common indication for HP-related lung transplantation in many centers.
Chemical Agents and Low-Molecular-Weight Haptens
- Isocyanates — Toluene diisocyanate (TDI), methylene diphenyl diisocyanate (MDI) — spray painters, polyurethane foam manufacturing. These chemicals act as haptens, binding to self-proteins to become immunogenic.
- Trimellitic anhydride (TMA) — Plastics and epoxy resin manufacturing.
- Metalworking fluids — Mycobacterium immunogenum and other organisms in coolant fluids; Metalworking Fluid HP increasingly recognized in machining industries.
Plant and Vegetable Dusts
- Bagassosis — Thermophilic actinomycetes from moldy sugar cane (bagasse) in paper and wallboard manufacturing.
- Mushroom Worker's Lung — Thermophilic bacteria and fungal spores in mushroom compost.
- Coffee Worker's Lung — Unroasted green coffee bean proteins.
- Grain dust — Grain Handler's Disease; multiple antigens including fungal spores and storage mites.
- Suberosis — Cork dust (Penicillium frequentans) in cork workers.
Pathophysiology — Immune Mechanisms
HP is the result of an aberrant immune response in a genetically susceptible host after repeated antigen inhalation. Two immunological pathways operate in sequence and in parallel:
Type III Hypersensitivity — Immune Complex Phase
On early antigen exposure, the immune system generates precipitating IgG antibodies (precipitins) directed against the inhaled antigen. Subsequent exposures lead to antigen-antibody immune complex formation in the alveolar walls and small airways. These complexes activate the complement cascade (C3a, C5a anaphylatoxins), attracting neutrophils within 4–8 hours of exposure. This accounts for the acute-phase symptoms — fever, chills, and dyspnea appearing hours after exposure and resolving within 12–48 hours when exposure stops.
Type IV Hypersensitivity — T-Cell-Mediated Phase
The delayed hypersensitivity component is orchestrated by antigen-presenting alveolar macrophages activating CD4+ T helper 1 (Th1) cells and CD8+ cytotoxic T lymphocytes. Th1 cytokines (IFN-γ, TNF-α, IL-12) drive macrophage activation, leading to the formation of noncaseating granulomas — found in approximately 70% of HP biopsy specimens. These granulomas are poorly formed and loosely organized, distinguishing them from the compact granulomas of sarcoidosis.
Persistent antigen stimulation shifts the balance toward fibrogenic pathways: activated macrophages and T cells secrete TGF-β and PDGF, driving fibroblast proliferation and collagen deposition. This progression from inflammation to fibrosis is the critical, potentially irreversible turning point in chronic HP.
Bronchoalveolar Lavage Immunology
BAL fluid in active HP is characterized by striking lymphocytosis — typically >20% and often 40–80% of cells. The CD4:CD8 ratio is typically low (<1.0, often <0.5), reflecting CD8+ cytotoxic T-cell predominance. This pattern contrasts with sarcoidosis, where CD4:CD8 ratios are elevated (>3.5), and helps distinguish the two conditions. Mast cells are increased in Bird Fancier's Lung. Neutrophilia is more prominent in Farmer's Lung and in acute-phase HP.
Why Only Some Exposed Individuals Develop HP
Genetic susceptibility is central. HLA alleles (particularly HLA-DPB1) determine the efficiency of antigen presentation to T cells. Gene polymorphisms in innate immune receptors (TLR9, NOD2) and in cytokine networks (TNF-α promoter) influence the magnitude and character of the inflammatory response. Environmental cofactors — antigen dose, particle size (respirable particles 0.5–5 microns deposit in alveoli), and concurrent respiratory infections — also modulate risk.
The paradoxical protection of smoking in acute HP relates to nicotine-mediated suppression of alveolar macrophage antigen presentation and reduced precipitin production. However, this protection is lost in chronic HP, where smoking accelerates fibrotic progression — making smoking cessation essential for all HP patients.
Clinical Phases — Acute, Subacute, Chronic
HP presents across a clinical spectrum determined by the intensity and duration of antigen exposure. The three clinical phases are not always distinct — patients may skip the acute phase entirely and present with subacute or chronic disease.
Acute HP
The classic presentation follows 4–8 hours after a heavy, discrete antigen exposure (for example, working in a barn after opening a moldy hay bale):
- Abrupt fever, chills, and rigors
- Myalgia and profound malaise
- Dry, nonproductive cough
- Dyspnea, chest tightness
- Bilateral fine inspiratory crackles on auscultation
- Hypoxemia in moderate-to-severe cases
Symptoms typically resolve within 12–48 hours after removal from the antigen source — a hallmark that distinguishes HP from other causes of fever and dyspnea. With repeated exposures, episodes recur reliably with each antigen contact. This pattern is frequently misdiagnosed as recurrent pneumonia or influenza for months or years before the exposure pattern is recognized. Chest X-ray may show bilateral, patchy ground-glass opacities or be normal.
Subacute HP
Results from lower-level but sustained or recurrent antigen exposure over weeks to months. Onset is insidious:
- Progressive exertional dyspnea
- Productive cough
- Fatigue and unintentional weight loss
- Bilateral crackles; early clubbing in some patients
- Chest X-ray: bilateral patchy or diffuse infiltrates
Subacute HP occupies a critical therapeutic window — antigen avoidance at this stage, with or without corticosteroids, usually results in substantial or complete recovery before fibrosis becomes established.
Chronic HP
The result of prolonged low-level antigen exposure, or the cumulative effect of repeated acute episodes without adequate antigen removal. This is the most clinically challenging phase:
- Progressive, often severe dyspnea on exertion and at rest
- Prominent digital clubbing (more common than in acute/subacute HP)
- Weight loss, anorexia
- Bilateral coarse crackles; wheeze in some patients
- Restrictive physiology on pulmonary function tests: reduced FVC, TLC, and DLCO
- Honeycombing and traction bronchiectasis on HRCT in advanced cases
Chronic HP can closely mimic Usual Interstitial Pneumonia (UIP)/Idiopathic Pulmonary Fibrosis (IPF) — both clinically and radiologically. This distinction matters enormously because IPF does not respond to corticosteroids, while active inflammatory HP may. Key distinguishing features: upper-lobe predominant fibrosis (vs. lower-lobe in IPF), antigen exposure history, BAL lymphocytosis, and a UIP pattern on biopsy without HP histological features (poorly-formed granulomas, peribronchiolar accentuation).
In the chronic fibrotic phase, disease may progress even after complete antigen avoidance due to autoimmune self-perpetuation of fibrosis, analogous to IPF. Median survival from the time fibrosis is established is approximately 5–7 years.
Diagnosis — Imaging, BAL, and Biopsy
No single test diagnoses HP definitively. Diagnosis requires integrating antigen exposure history, clinical features, HRCT findings, BAL results, serum precipitins, and sometimes surgical lung biopsy. International guidelines (Raghu et al., 2020) provide a structured diagnostic framework using these components.
High-Resolution CT (HRCT)
HRCT is the single most informative non-invasive test. Patterns vary by phase:
- Acute/Subacute HP:
- Diffuse bilateral ground-glass opacities (hazy increased attenuation that does not obscure underlying vessels)
- Poorly defined centrilobular nodules (2–4 mm) — reflecting peribronchiolar inflammation
- Mosaic attenuation — geographic areas of different lung density reflecting heterogeneous small airway involvement
- Expiratory air trapping — lobular or geographic lucent areas on expiratory CT; a highly characteristic and sensitive finding for HP even when inspiratory CT appears near-normal
- Chronic Fibrotic HP:
- Upper-lobe predominant fibrosis — important distinguisher from IPF (lower-lobe predominant)
- Traction bronchiectasis and bronchiolectasis
- Honeycombing (less common than in IPF)
- Relative sparing of the lung bases compared to UIP/IPF
- "Three-density sign" (highly characteristic): Simultaneous presence of ground-glass attenuation (active inflammation), normal-density lobules, and lobular air trapping on expiratory images — creates a geographic patchwork pattern strongly suggesting HP.
Bronchoalveolar Lavage (BAL)
BAL is central to HP diagnosis and should be performed when HP is suspected. Key findings:
- Lymphocytosis >20% (normal <15%); often 40–80% in active acute or subacute HP
- Low CD4:CD8 ratio (<1.0, typically <0.5) — CD8+ cytotoxic T-cell predominance distinguishes HP from sarcoidosis (CD4:CD8 >3.5)
- BAL lymphocytosis >30% is a strong positive predictor for HP diagnosis
- Mast cells elevated in Bird Fancier's Lung
- Plasma cells may be present
- Neutrophilia more prominent in Farmer's Lung and acute-phase HP
BAL lymphocytosis is not specific to HP — it also occurs in sarcoidosis, nonspecific interstitial pneumonia (NSIP), and drug reactions — but in the context of exposure history and CT findings, it powerfully supports the diagnosis.
Serum Precipitins (Specific IgG)
Precipitating IgG antibodies against the suspected antigen are present in 40–90% of HP patients, depending on the antigen panel used and the laboratory method. Key considerations:
- Positive precipitins confirm antigen exposure and sensitization but do not diagnose HP — 15–50% of asymptomatic exposed farmers and bird keepers also have positive precipitins
- Negative precipitins do not exclude HP, especially if the specific antigen is not in the test panel
- Panels are available for: Farmer's Lung antigens (thermophilic actinomycetes), bird proteins (pigeon, budgerigar, parrot), Aspergillus species, MAC, and others
- Clinical utility: positive precipitins in the right clinical and CT context significantly increase diagnostic confidence and reduce need for biopsy
Pulmonary Function Tests (PFTs)
PFTs demonstrate the physiological impact and guide severity assessment:
- Restrictive pattern: Reduced FVC, TLC, and DLCO (carbon monoxide diffusing capacity)
- DLCO is the most sensitive PFT measure — reduced even before spirometric restriction appears
- Obstructive or mixed pattern may be present, especially in Farmer's Lung and Hot Tub Lung
- Serial PFTs every 3–6 months are used to monitor treatment response and disease progression
Specific Inhalation Challenge (SIC)
The reference standard for occupational HP. The patient is exposed to controlled concentrations of the suspected antigen and monitored for physiological and symptomatic responses. Used when the causative antigen is unclear or when occupational compensation claims require objective documentation. Performed only in specialized centers.
Surgical Lung Biopsy (VATS)
Video-assisted thoracoscopic surgery (VATS) biopsy is the gold standard for atypical, chronic, or diagnostically uncertain HP — particularly when the CT pattern overlaps with UIP/IPF. Characteristic histology:
- Peribronchiolar lymphocytic infiltration — cellular infiltrate centered on terminal bronchioles; airway-centered rather than purely alveolar
- Poorly-formed noncaseating granulomas (~70% of HP biopsies) — loosely organized, often with giant cells; less compact than sarcoid granulomas
- Organizing pneumonia component
- In chronic HP: bridging fibrosis, honeycomb change; UIP-like pattern may be present, requiring careful correlation with clinical and CT findings to distinguish from IPF
Diagnostic Scoring Frameworks
International guidelines (Raghu et al., 2020) use a structured probability-based approach combining CT pattern (typical, compatible, indeterminate), BAL lymphocytosis, precipitins, and antigen exposure to classify HP probability as high, intermediate, or low — reducing the need for surgical biopsy in high-probability cases. This framework is increasingly used in clinical practice.
Treatment — Antigen Avoidance and Corticosteroids
1. Antigen Identification and Avoidance
Antigen avoidance is the cornerstone of HP management and the single most effective intervention. A thorough occupational and environmental history — ideally with a home visit or workplace inspection — is essential to identify and eliminate the causative exposure.
- Farmer's Lung: Switch to mechanically dried hay; use positive-pressure helmets or powered air-purifying respirators (PAPRs) when avoidance is impossible; improved barn ventilation
- Bird Fancier's Lung: Complete removal of birds from home and workplace is the most effective measure; N95 respirators alone are insufficient for high-risk exposures; decontaminate living areas (bird antigen persists in carpets and upholstery for months to years after bird removal)
- Hot Tub Lung: Eliminate the indoor hot tub, or shock-chlorinate and maintain strict water treatment; improved ventilation
- Occupational HP: Relocation away from the exposure area when engineering controls are insufficient; disability assessment if respiratory symptoms preclude continued employment
In acute and subacute HP, antigen removal alone may result in complete clinical and radiological recovery. In chronic fibrotic HP, fibrosis can continue to progress even after complete antigen avoidance due to self-sustaining fibrogenic mechanisms — this is the critical argument for early diagnosis and intervention.
2. Corticosteroids
Corticosteroids are the primary pharmacological treatment for active HP:
- Acute HP: Prednisone 40–60 mg/day (or equivalent) for 1–2 weeks, tapered over 4–8 weeks as symptoms and PFTs improve. Accelerates recovery.
- Subacute HP: Similar dosing; taper guided by clinical and PFT response. Most patients respond well if antigen avoidance is concurrent.
- Chronic HP: Evidence is mixed. The ISAM study (Rose et al., 2020) — a randomized controlled trial — found no significant benefit of prednisolone over placebo in patients with chronic HP over 52 weeks in terms of FVC change or survival. Despite this, corticosteroids remain widely used in practice for active inflammatory chronic HP, particularly when BAL lymphocytosis and ground-glass opacity suggest ongoing inflammation rather than pure fibrosis.
3. Steroid-Sparing Immunosuppressants
For patients with steroid-refractory, steroid-dependent, or progressive chronic HP who cannot tolerate long-term prednisone:
- Azathioprine (1.5–2 mg/kg/day) — most commonly used steroid-sparing agent; monitor CBC and LFTs
- Mycophenolate mofetil (MMF) (1000–1500 mg twice daily) — increasingly used; may be better tolerated than azathioprine
- Duration typically 12–24 months; taper with PFT monitoring
4. Antifibrotic Therapy
A landmark advance came with the INBUILD trial (Flaherty et al., 2019), which demonstrated that nintedanib — a tyrosine kinase inhibitor targeting PDGFR, VEGFR, and FGFR — significantly slowed the rate of FVC decline in patients with progressive fibrosing ILDs other than IPF, including fibrotic HP. This led to regulatory approval of nintedanib for progressive fibrosing ILDs in multiple countries.
- Nintedanib 150 mg twice daily — indicated for fibrotic HP with documented FVC decline; main side effects: diarrhea, nausea, hepatotoxicity (monitor LFTs)
- Does not replace antigen avoidance or immunosuppression — used as adjunct in progressive fibrotic HP
5. Lung Transplantation
Lung transplantation is the definitive treatment for end-stage fibrotic HP:
- Indicated when FVC falls below 50% predicted, DLCO below 35%, or there is rapid physiological decline despite maximal medical therapy
- Bird Fancier's Lung is one of the most common indications for HP-related lung transplantation
- Post-transplant outcomes are comparable to other ILD indications; antigen avoidance must continue post-transplant to prevent recurrence in the allograft
- Early referral to transplant centers is recommended once significant fibrosis is detected
6. Monitoring
- Pulmonary function tests (FVC, TLC, DLCO) every 3–6 months during active treatment
- 6-minute walk test and oxygen saturation assessment
- HRCT annually or when clinical status changes
- Echocardiogram to screen for pulmonary hypertension in advanced disease
Prognosis
Prognosis in HP depends heavily on the clinical phase at diagnosis, the specific causative antigen, and whether antigen avoidance is achieved:
Acute and Subacute HP
The outlook is generally excellent with prompt antigen avoidance. Most patients recover completely or near-completely when the antigen source is removed early. PFTs normalize within weeks to months. The key prognostic variable is avoidance: those who continue exposure despite diagnosis have significantly worse outcomes.
Chronic Fibrotic HP
Prognosis is considerably worse once established fibrosis is present:
- Median survival from fibrosis diagnosis: 5–7 years
- 5-year mortality: approximately 30%
- Bird Fancier's Lung carries a worse prognosis than Farmer's Lung — partly because bird antigen is more difficult to completely eliminate from the home environment
Predictors of Poor Prognosis
- Honeycombing or UIP-like pattern on HRCT
- UIP histological pattern on biopsy without HP-specific features
- FVC <60% predicted at presentation
- DLCO <40% predicted
- Continued antigen exposure despite diagnosis
- Older age at diagnosis
- Presence of pulmonary hypertension
- Rapid FVC decline (>10% relative decline per year)
Chronic HP vs. IPF
Distinguishing chronic fibrotic HP from IPF has direct prognostic and therapeutic implications. Chronic HP has a somewhat better median survival than IPF (5–7 years vs. 3–5 years for IPF), but the overlap is substantial. When clinical and radiological features are ambiguous, surgical biopsy is essential — misclassifying HP as IPF (and withholding antigen avoidance counseling) can accelerate otherwise preventable progression.
Prevention
Prevention of HP centers on controlling antigen exposure at the source before sensitization and clinical disease develop:
Agricultural Settings
- Mechanical drying of hay and grain to moisture content <15% before storage (eliminating thermophilic actinomycete growth)
- Improved barn and silo ventilation
- Powered air-purifying respirators (PAPRs) when working in high-dust areas
- Surveillance programs for farmers with recurrent respiratory illness
Bird Exposure
- Awareness campaigns for bird fanciers about the risk of Bird Fancier's Lung
- Routine health surveillance for pigeon breeders and aviary workers
- Handling birds outdoors when possible; washing hands and changing clothes after bird contact
Indoor Water Systems
- Regular maintenance and decontamination of indoor hot tubs and heated pools
- Adequate ventilation of indoor pool areas to disperse MAC aerosols
- Monitoring water quality (chlorine levels, pH)
Occupational Settings
- Engineering controls (local exhaust ventilation, enclosed systems) to reduce airborne antigen concentrations
- Pre-employment screening and periodic health surveillance for workers in high-risk industries (spray painting, foam manufacturing, metalworking)
- Substitution of high-risk chemicals (e.g., replacing TDI with less reactive isocyanates where feasible)
- N95 respirator use as a supplemental measure when engineering controls are insufficient
Early Diagnosis
The most impactful preventive measure at the individual level is early diagnosis before fibrosis develops. A high index of clinical suspicion — particularly in any patient with recurrent flu-like respiratory illness, unexplained interstitial infiltrates, or subacute dyspnea — combined with systematic antigen exposure inquiry can identify HP at a stage where complete recovery is still achievable.
Research Papers
Search PubMed for current research on hypersensitivity pneumonitis:
- Hypersensitivity pneumonitis extrinsic allergic alveolitis
- Farmer's lung thermophilic actinomycetes
- Bird fancier's lung avian antigen
- Hot tub lung mycobacterium avium complex
- Hypersensitivity pneumonitis CT mosaic attenuation
- Hypersensitivity pneumonitis bronchoalveolar lavage lymphocytosis
- Hypersensitivity pneumonitis antigen avoidance
- Hypersensitivity pneumonitis corticosteroid treatment
- Chronic hypersensitivity pneumonitis fibrosis nintedanib
- Hypersensitivity pneumonitis vs IPF differential diagnosis
- HP diagnosis serum precipitins IgG
- Hypersensitivity pneumonitis lung transplantation
References
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
Connections
- Pulmonology
- Sarcoidosis
- Interstitial Lung Disease
- Asthma
- COPD
- Pulmonary Hypertension
- Pneumonia
- Rheumatoid Arthritis
- Lung Cancer
- Tuberculosis
- Allergies
- ARDS
- Mold Toxicity