Soft Tissue Sarcoma
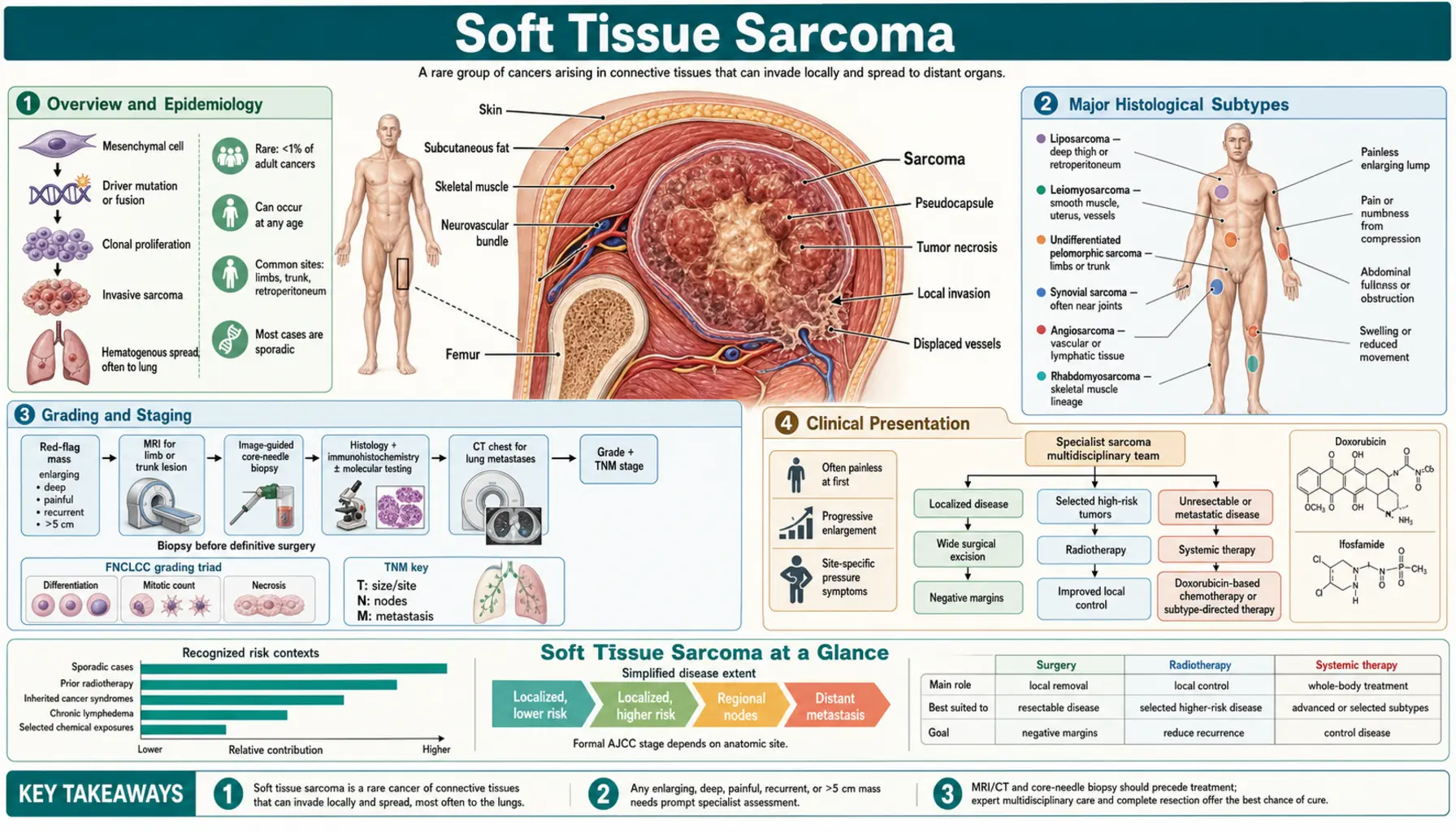
Soft tissue sarcomas (STS) are a diverse family of malignant tumors that arise from the connective and supportive tissues of the body — muscles, fat, blood vessels, nerves, tendons, and the tissue around joints. With more than 50 distinct histological subtypes recognized by the World Health Organization, they are among the most complex cancers a patient or physician can face. Although rare in adults (fewer than 1% of all adult malignancies), soft tissue sarcomas account for roughly 7 to 15 percent of childhood cancers. Each year approximately 13,000 new cases are diagnosed in the United States, and around 5,000 people die from the disease. Understanding the specific subtype, grade, and location of a sarcoma is essential, because treatment — and outlook — varies dramatically between subtypes.
- Overview and Epidemiology
- Major Histological Subtypes
- Grading and Staging
- Clinical Presentation
- Diagnosis
- Surgery
- Radiation Therapy
- Systemic Therapy — Advanced Disease
- Histotype-Specific Approaches
- Prognosis and Surveillance
- Key Research Papers
- PubMed Topic Searches
- Connections
Overview and Epidemiology
Soft tissue sarcomas can arise anywhere in the body where connective tissue exists — which means virtually anywhere. The extremities (thighs and upper arms) are the most common primary site, accounting for about 43 percent of cases. The retroperitoneum (the space behind the abdominal cavity) accounts for roughly 15 percent, and the remaining cases arise in the trunk, head and neck, and visceral organs. Retroperitoneal sarcomas present a particular challenge because they often grow very large before causing symptoms, and the anatomy limits the ability to achieve clean surgical margins.
The incidence of soft tissue sarcoma rises with age, with the median age at diagnosis around 58 years for most adult subtypes. However, certain subtypes — rhabdomyosarcoma and synovial sarcoma — predominantly affect children and young adults. Risk factors include prior radiation therapy (radiation-induced sarcomas typically appear 10 years or more after treatment), certain hereditary syndromes (Li-Fraumeni syndrome with TP53 germline mutations, neurofibromatosis type 1, familial retinoblastoma with RB1 loss), chronic lymphedema (predisposing to angiosarcoma), and occupational exposure to vinyl chloride (associated with hepatic angiosarcoma). In most patients, however, no clear cause is found.
Because of their rarity and complexity, soft tissue sarcomas should be managed at specialized sarcoma centers. Studies consistently show that patients treated at high-volume centers by multidisciplinary sarcoma teams have better outcomes, lower local recurrence rates, and are more likely to achieve limb-sparing surgery rather than amputation.
Major Histological Subtypes
The WHO Classification of Soft Tissue and Bone Tumors recognizes over 50 malignant soft tissue subtypes. Each has distinct molecular drivers, preferred anatomic locations, and treatment sensitivities. The most clinically important subtypes include:
Liposarcoma
The most common adult soft tissue sarcoma, arising from adipose tissue. There are three main subtypes with very different biology. Well-differentiated and dedifferentiated liposarcoma share amplification of chromosome 12q13-15, which drives overexpression of MDM2 (an inhibitor of the tumor-suppressor p53) and CDK4. Well-differentiated tumors are locally aggressive but rarely metastasize; dedifferentiated tumors have a component that has lost fat differentiation and behaves more aggressively, with metastatic rates around 15 to 20 percent. MDM2 FISH or immunohistochemistry is the key diagnostic test. Myxoid/round cell liposarcoma harbors the pathognomonic FUS-DDIT3 fusion (from the t(12;16)(q13;p11) translocation), or less commonly EWSR1-DDIT3. Myxoid LPS is particularly sensitive to trabectedin and to radiation. Pleomorphic liposarcoma is the rarest and most aggressive subtype, with complex karyotypes and poor prognosis.
Leiomyosarcoma
A malignant tumor of smooth muscle. Leiomyosarcomas arise most commonly in the uterus, the retroperitoneum (often from large retroperitoneal vessels), and from vascular walls throughout the body. Uterine leiomyosarcoma is distinct from the far more common uterine leiomyoma (fibroid) and carries a poor prognosis when advanced. Vascular leiomyosarcoma of the inferior vena cava presents unique surgical challenges. Leiomyosarcoma is among the subtypes that respond relatively well to gemcitabine plus docetaxel chemotherapy in the second-line setting.
Undifferentiated Pleomorphic Sarcoma (UPS)
Previously called malignant fibrous histiocytoma (MFH), UPS is a diagnosis of exclusion — a high-grade sarcoma that shows no line of differentiation even after thorough pathologic workup including immunohistochemistry and molecular testing. It is one of the most common adult STS subtypes and arises most frequently in the extremities of older adults. Prognosis is poor for high-grade disease.
Synovial Sarcoma
Despite its name, synovial sarcoma does not arise from synovium and can occur in locations far from joints. It primarily affects young adults between 15 and 40 years of age and is the second most common STS in adolescents and young adults. The molecular hallmark is the SS18-SSX1 or SS18-SSX2 fusion from the t(X;18)(p11;q11) translocation. Synovial sarcoma is one of the most chemotherapy-sensitive sarcoma subtypes — doxorubicin and ifosfamide produce objective responses in 40 to 60 percent of patients with advanced disease. Emerging data support NY-ESO-1-targeting T-cell therapies.
Gastrointestinal Stromal Tumor (GIST)
GISTs arise from interstitial cells of Cajal in the gastrointestinal tract (most commonly the stomach and small intestine). They are driven by activating mutations in KIT (approximately 80%) or PDGFRA (approximately 10%), or rarely by SDH, NF1, or BRAF alterations. GISTs are treated as a separate entity from other STS because they are largely resistant to conventional sarcoma chemotherapy but exquisitely sensitive to the tyrosine kinase inhibitor imatinib (Gleevec). GISTs represent one of the landmark success stories of molecularly targeted therapy in oncology.
Rhabdomyosarcoma
The most common soft tissue sarcoma in children, representing about 50 percent of pediatric STS. Embryonal rhabdomyosarcoma (ERMS), which is the more favorable subtype, typically occurs in children under 10 and frequently involves the head, neck, and genitourinary tract. Alveolar rhabdomyosarcoma (ARMS) carries FOXO1 gene fusions — PAX3-FOXO1 or PAX7-FOXO1 — and has a worse prognosis. Treatment combines surgery, radiation, and multiagent chemotherapy.
Angiosarcoma
An aggressive vascular malignancy with three main forms: cutaneous angiosarcoma of the scalp and face (predominantly elderly men), post-mastectomy lymphedema-associated angiosarcoma (Stewart-Treves syndrome), and radiation-induced angiosarcoma appearing years after breast radiation. Angiosarcoma is highly vascular, bleeds easily, and is often multifocal. It carries one of the poorest prognoses among STS. Gemcitabine, taxanes, and anti-angiogenic agents (bevacizumab, sorafenib) are used in the systemic setting.
Epithelioid Sarcoma and Clear Cell Sarcoma
Epithelioid sarcoma is a rare tumor most common in the distal extremities of young adults, characterized by loss of the SMARCB1/INI1 tumor suppressor through deletion or mutation; this loss makes it potentially targetable by EZH2 inhibitors (tazemetostat). Clear cell sarcoma, sometimes called "melanoma of soft parts," harbors EWSR1-ATF1 fusions, produces melanin, and expresses melanocytic markers (S100, HMB-45). Despite the molecular resemblance to melanoma, it does not respond to BRAF or checkpoint inhibitor therapies in the same way.
Grading and Staging
Histological grade is the single most important prognostic factor for soft tissue sarcoma. The FNCLCC (French Federation of Cancer Centers Sarcoma Group) grading system is the international standard and assigns scores in three categories:
- Tumor differentiation (1–3 points): how closely the tumor resembles normal tissue
- Mitotic rate (1–3 points): the number of cell divisions seen per 10 high-power microscopic fields
- Tumor necrosis (0–2 points): the fraction of the tumor that is dead tissue
Scores are summed to assign grade: Grade 1 (2–3 points, low-grade), Grade 2 (4–5 points, intermediate-grade), Grade 3 (6–8 points, high-grade). Low-grade sarcomas rarely metastasize; high-grade tumors have metastatic risk of 30 to 50 percent or more.
The AJCC TNM staging system (8th edition) adds anatomic extent. Tumor size thresholds are T1 (5 cm or less), T2 (greater than 5 cm to 10 cm), T3 (greater than 10 cm to 15 cm), and T4 (greater than 15 cm). Depth relative to the investing fascia (superficial vs. deep) was removed from the 8th edition. Regional lymph node involvement (N1) and distant metastasis (M1) complete staging. It is important to note that retroperitoneal sarcoma staging and prognostic nomograms differ from extremity staging because tumor size limits are less meaningful when tumors commonly exceed 20 to 30 cm at presentation.
Clinical Presentation
The most common presentation of soft tissue sarcoma is a painless, slowly enlarging mass. This is precisely what makes early diagnosis so difficult — a mass that does not hurt is easy to dismiss as a benign lipoma or cyst. Any soft tissue mass that is larger than 5 cm, deep to the investing fascia of a muscle, growing rapidly, or recurring after previous excision should be considered a sarcoma until proven otherwise and warrants urgent imaging and biopsy at a specialist center.
For extremity sarcomas, the thigh is the most common location. Patients often report first noticing the mass weeks to months before seeking care, or discover it incidentally. Pain is present in roughly one-third of cases and usually reflects pressure on nearby nerves or bones.
For retroperitoneal sarcomas, the presentation is typically a large abdominal or flank mass causing vague abdominal fullness, early satiety, or back pain. Because the retroperitoneum accommodates enormous tumors before compressing vital structures, retroperitoneal sarcomas are frequently discovered at very large sizes — often 10 to 30 cm. Early detection requires imaging for an unrelated reason.
The most common site of distant metastasis for most STS subtypes is the lungs. Nodal metastasis is rare (around 3 to 5%) for most subtypes but is more common in rhabdomyosarcoma, epithelioid sarcoma, clear cell sarcoma, and angiosarcoma. Bone and liver metastases occur in advanced disease. Myxoid liposarcoma has an unusual predilection for unusual sites including the spine and retroperitoneum.
Diagnosis
Correct diagnosis of a soft tissue sarcoma requires a specific sequence of steps, and getting that sequence wrong can compromise both diagnosis and treatment.
MRI is the gold standard for local staging and must be performed before biopsy. MRI characterizes the tumor's relationship to neurovascular structures, fascial planes, and bone — information essential for surgical planning. Contrast-enhanced MRI sequences including T1, T2, and fat-suppressed post-gadolinium images provide the best tissue characterization.
Never biopsy before MRI. Biopsy changes tissue planes, can seed tumor cells along the needle track, and alters MRI signal in ways that make subsequent imaging less interpretable. The biopsy tract must be planned so it can be excised en bloc with the tumor at definitive surgery.
The preferred biopsy technique is core needle biopsy (CNB), ideally ultrasound-guided. Multiple cores (typically 3 to 6) should be obtained. The skin entry site should be placed in line with the planned surgical incision, not transversely across the extremity (a transverse incision would require a more extensive field to excise the contaminated tract). Fine needle aspiration (FNA) alone provides insufficient material for the immunohistochemical and molecular studies needed to classify sarcoma. Incisional biopsy in the operating room is reserved for cases where CNB is non-diagnostic.
Pathology workup includes hematoxylin-and-eosin staining, a comprehensive immunohistochemistry panel (vimentin, desmin, SMA, S100, MDM2, CD34, CD117/KIT, DOG-1, TLE1 for synovial sarcoma, SMARCB1/INI1), and molecular studies. FISH is used to detect gene amplification (MDM2 in WD/DDLPS) and translocations (SS18-SSX in synovial sarcoma, FUS-DDIT3 in myxoid LPS, EWSR1 fusions). Next-generation sequencing (NGS) panels are increasingly standard, both for diagnosis and to identify actionable mutations.
For staging, CT of the chest is required for all patients with intermediate- or high-grade sarcoma to evaluate for pulmonary metastases. CT of the abdomen and pelvis is appropriate for retroperitoneal or visceral sarcomas. PET-CT provides limited additional staging information for most subtypes but may be useful in rhabdomyosarcoma and for detecting nodal disease.
Surgery
Surgery remains the cornerstone of curative treatment for localized soft tissue sarcoma. The goal is wide local excision with negative (R0) margins — a rim of normal tissue surrounding the tumor on all sides. Even a microscopic positive margin (R1) substantially increases local recurrence risk.
Limb-sparing surgery is now achievable in approximately 90 percent of extremity sarcoma patients treated at specialized centers, where experienced sarcoma surgeons can reconstruct vessels, nerves, and bone when needed. Amputation is reserved for the rare cases where adequate margins cannot be achieved without sacrificing the limb's function, or where reconstruction is not possible. Limb-sparing surgery combined with radiation therapy provides equivalent local control and survival to amputation while preserving the limb.
For retroperitoneal sarcomas, the standard approach is complete cytoreductive surgery — removal of the entire tumor along with any directly involved structures (kidney, colon, spleen, pancreatic tail). Because the retroperitoneum allows tumors to displace rather than invade adjacent organs, a skilled surgeon can often achieve complete resection even of very large tumors. Incomplete resection (debulking) provides limited benefit and is generally not recommended. The Italian Sarcoma Group and Trans-Atlantic Retroperitoneal Sarcoma Working Group (TARPSWG) have published detailed consensus guidelines for the surgical management of retroperitoneal STS.
Re-excision of inadequately excised sarcomas (when initial surgery was performed without prior imaging or biopsy at a non-specialist facility) should be performed at a specialized center, ideally before adjuvant treatment. Residual tumor is found in approximately 50 percent of such re-excision specimens.
Radiation Therapy
Radiation therapy is an integral component of treatment for most intermediate- and high-grade extremity sarcomas, and for selected retroperitoneal tumors. It reduces local recurrence rates from approximately 40 percent with surgery alone to approximately 10 to 15 percent with combined surgery and radiation.
The landmark Canadian National Cancer Institute (NCI) SR.2 randomized trial (O'Sullivan et al., 2002) compared preoperative (50 Gy in 25 fractions) versus postoperative (66 Gy in 33 fractions) radiation for extremity STS. The two approaches achieved equivalent local control and survival. The key differences are practical:
- Preoperative radiation uses a smaller radiation field (treats less normal tissue), takes advantage of better tumor oxygenation for radiosensitivity, may downsize tumors to facilitate surgery, and results in less long-term fibrosis and edema — but is associated with higher acute wound complication rates (approximately 35% vs. 17%).
- Postoperative radiation uses a larger field (must cover the entire surgical bed plus margin), and results in more chronic fibrosis, joint stiffness, and limb edema — but wound healing is complete before radiation begins, so acute wound problems are fewer.
Most specialized centers now prefer preoperative radiation for extremity sarcomas amenable to wound reconstruction, because the long-term functional benefits (less fibrosis) outweigh the short-term wound risk. Preoperative radiation is particularly favored when achieving clear margins is anticipated to be difficult.
For retroperitoneal sarcomas, the role of radiation remains controversial. The STRASS randomized trial (EORTC-STBSG) evaluated preoperative radiation in retroperitoneal STS but did not demonstrate an improvement in abdominal recurrence-free survival overall; subset analyses suggested possible benefit for well-differentiated liposarcoma histology. Postoperative radiation in the retroperitoneum is complicated by bowel toxicity from the large field required.
Systemic Therapy — Advanced and Metastatic Disease
For patients with unresectable locally advanced or metastatic soft tissue sarcoma, systemic chemotherapy is the primary treatment. Unfortunately, overall response rates are modest and prolonged remissions are uncommon for most subtypes.
First-line treatment: Doxorubicin (75 mg/m² every 3 weeks) is the standard first-line backbone for most STS subtypes. Adding ifosfamide to doxorubicin increases response rates (25–35% vs. 14% for doxorubicin alone) but also increases toxicity (myelosuppression, nephrotoxicity, encephalopathy) and has not consistently improved overall survival in randomized trials. Combination therapy is generally reserved for patients with good performance status in whom tumor shrinkage is a meaningful goal (e.g., to render an initially unresectable tumor resectable).
Gemcitabine plus docetaxel is a widely used second-line (and sometimes first-line) regimen with particular activity in leiomyosarcoma and angiosarcoma (response rates of approximately 17–20% in the second-line setting).
Trabectedin (Yondelis) was FDA-approved in 2015 for unresectable or metastatic liposarcoma or leiomyosarcoma after failure of anthracycline-based chemotherapy, based on the TRAILBLAZER trial (Demetri et al., 2016). It showed improvement in progression-free survival (4.2 vs. 1.5 months) over dacarbazine, though objective response rates were modest (9.9%). It is particularly active in myxoid liposarcoma.
Pazopanib (Votrient), a multi-targeted tyrosine kinase inhibitor, was approved in 2012 for non-adipocytic soft tissue sarcomas after failure of prior chemotherapy, based on the PALETTE trial (van der Graaf et al., 2012). It improved median progression-free survival from 1.6 to 4.6 months versus placebo. It is not indicated for liposarcoma (specifically excluded from PALETTE due to lack of efficacy).
Eribulin (Halaven) was approved by the FDA in 2016 for unresectable or metastatic liposarcoma after failure of at least one prior anthracycline-based regimen, based on the Schoffski et al. Lancet 2016 trial, which demonstrated a survival benefit (13.5 vs. 11.5 months median OS) over dacarbazine.
Olaratumab lesson: The PDGFR-alpha antibody olaratumab initially received accelerated approval in 2016 based on a phase 2 trial showing dramatic improvement in overall survival with doxorubicin (Tap et al., 2016). However, the confirmatory phase 3 ANNOUNCE trial (Tap et al., 2020) failed to show benefit, and olaratumab was voluntarily withdrawn from the market in 2019 — an important reminder that phase 2 results in sarcoma must be interpreted with caution.
Imatinib for GIST: For GIST, imatinib (400 mg daily) is the standard of care in the first-line metastatic and adjuvant setting. Second-line options after imatinib progression include sunitinib, and third-line regorafenib or ripretinib.
Histotype-Specific Systemic Approaches
The era of "one chemotherapy fits all sarcomas" is over. Histotype-specific treatment selection is now the standard of care at specialized centers:
- Synovial sarcoma: High sensitivity to doxorubicin plus ifosfamide (response rates 40–60%). High-dose ifosfamide (14 g/m² over 14 days) may achieve responses in doxorubicin-pretreated patients. NY-ESO-1 TCR T-cell therapy (afami-cel/ADP-A2M4) has shown promising response rates in NY-ESO-1-positive synovial sarcoma.
- Rhabdomyosarcoma: VAC (vincristine, actinomycin D, cyclophosphamide) backbone chemotherapy with radiation for non-metastatic disease achieves 5-year survival rates of 70 to 90% for low-risk and intermediate-risk groups. Alveolar histology with FOXO1 fusion is a high-risk feature.
- Myxoid liposarcoma: Trabectedin produces objective responses in 24 to 50% of patients and is considered a preferred first or second-line agent. Radiation sensitivity is high; pre-op radiation is particularly effective for myxoid LPS.
- GIST: Imatinib 400 mg/day is standard; 800 mg/day may benefit KIT exon 9 mutations. The ACOSOG Z9001 and SSGXVIII trials established 3 years of adjuvant imatinib as standard after resection of high-risk localized GIST, improving recurrence-free survival (DeMatteo et al., 2009). Mutation genotyping is mandatory before starting therapy — PDGFRA D842V mutation is resistant to imatinib but sensitive to avapritinib.
- Angiosarcoma: Weekly paclitaxel or gemcitabine are active agents; bevacizumab and sorafenib have shown activity. Anti-angiogenic strategies reflect the vascular biology of this tumor.
- Epithelioid sarcoma: EZH2 inhibitor tazemetostat received FDA approval in 2020 for metastatic or locally advanced epithelioid sarcoma with SMARCB1/INI1 loss.
Prognosis and Surveillance
Prognosis in soft tissue sarcoma depends most powerfully on histological grade, tumor size, depth, and presence of metastatic disease at diagnosis. Anatomic site also matters — retroperitoneal tumors tend to have worse outcomes than extremity tumors of comparable grade and size, largely because of the difficulty achieving complete resection and the high local recurrence rate.
Approximate 5-year survival rates by stage:
- Low-grade (G1), any size, no metastases: approximately 85 to 90%
- High-grade (G3), T1 (≤5 cm), no metastases: approximately 70 to 80%
- High-grade (G3), T2–T4, no metastases: approximately 50 to 60% for extremity; worse for retroperitoneal
- Metastatic disease (any grade): approximately 15 to 20% at 5 years; median survival 12 to 18 months
Patients who achieve complete resection of limited pulmonary metastases (pulmonary metastaectomy) have better outcomes, with some reports of long-term survivors. This is considered standard practice at specialized centers for patients with controlled primary tumors and resectable lung-only disease.
Surveillance after definitive treatment is critical because local recurrence and lung metastases are most common in the first 2 to 3 years. Standard practice for high-grade STS includes:
- Clinical examination and MRI of the primary site every 3 to 4 months for the first 2 to 3 years, then every 6 months to 5 years, then annually
- CT of the chest every 3 to 6 months for the first 2 to 3 years for intermediate- and high-grade tumors
- Low-grade tumors may be followed less intensively (CT chest every 6 to 12 months)
Rehabilitation — including physical therapy, lymphedema management, and psychosocial support — is an essential component of post-treatment care, particularly after limb-sparing surgery and radiation.
Key Research Papers
- Casali PG et al. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2018;29(Suppl 4):iv51–iv67. — Search PubMed
- Gronchi A et al. Retroperitoneal sarcomas: present and future. Eur J Cancer 2017;86:75–85. — Search PubMed
- Sleijfer S et al. Pazopanib plus best supportive care versus placebo plus best supportive care in patients with relapsed or refractory soft-tissue sarcoma after failure of standard chemotherapy (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–1886. PMID 22595799
- Demetri GD et al. Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: results of a randomized phase III study of trabectedin versus dacarbazine (TRAILBLAZER). J Clin Oncol 2016;34:786–793. PMID 26371143
- Schoffski P et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet 2016;387:1629–1637. PMID 26874885
- van der Graaf WT et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86. PMID 22595799
- Tap WD et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–497. PMID 27291997
- Tap WD et al. Effect of doxorubicin-based chemotherapy on the prognosis of patients with advanced uterine leiomyosarcoma and soft tissue sarcoma (ANNOUNCE): a randomised, double-blind, multicentre, phase 3 trial. J Clin Oncol 2020;38:1701–1709. — Search PubMed
- O'Sullivan B et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 2002;359:2235–2241. PMID 12103287
- DeMatteo RP et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:1097–1104. PMID 19303137
- Gronchi A et al. Primary retroperitoneal sarcomas: a qualitative systematic review. Ann Surg 2009;250:697–707. — Search PubMed
- Linch M et al. Soft-tissue sarcomas — adult-type tumours. Lancet 2013;381:770–779. — Search PubMed
PubMed Topic Searches
- Soft tissue sarcoma treatment
- Liposarcoma chemotherapy
- Leiomyosarcoma systemic therapy
- Synovial sarcoma molecular biology
- GIST imatinib adjuvant therapy
- Retroperitoneal sarcoma surgery
- Rhabdomyosarcoma children treatment
- Sarcoma preoperative radiation therapy
- Angiosarcoma treatment outcomes
- Soft tissue sarcoma FNCLCC grading
Connections
- Oncology
- Cancer
- Sarcoma Overview
- Ewing Sarcoma
- Osteosarcoma
- Gastrointestinal Stromal Tumor
- Neuroblastoma
- Skin Cancer
- Lymphoma
- Neuroendocrine Tumors
- Kidney Cancer
- Lung Cancer