Gastrointestinal Stromal Tumor (GIST)
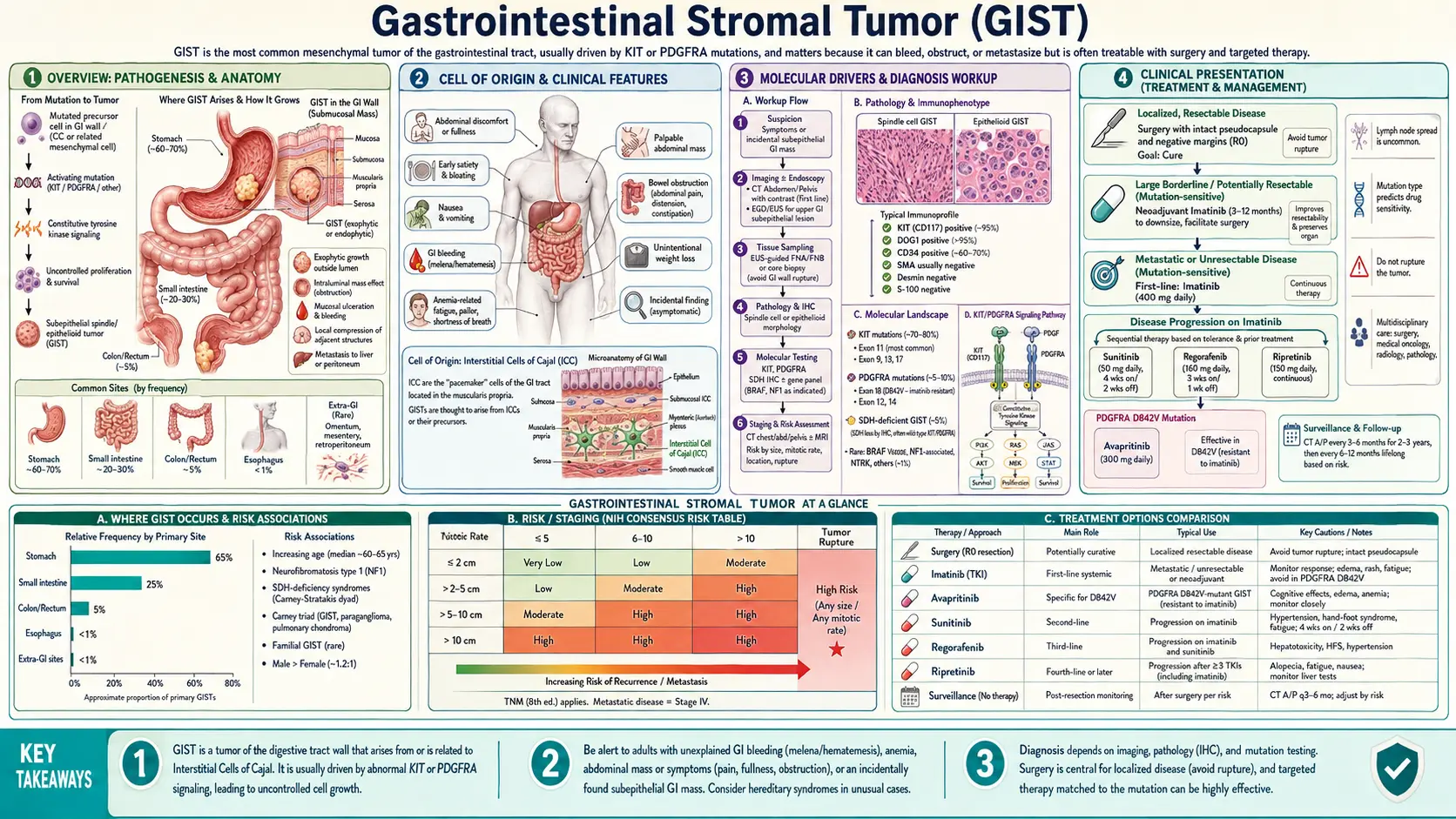
Gastrointestinal stromal tumor (GIST) is the most common mesenchymal tumor of the gastrointestinal tract, arising from the interstitial cells of Cajal — the pacemaker cells that coordinate GI motility. Once considered uniformly fatal in advanced disease, GIST was transformed into a manageable chronic condition by the discovery of its molecular driver (KIT mutations) and the introduction of imatinib mesylate (Gleevec) in 2001 — one of the first and most dramatic examples of targeted cancer therapy in oncology. Today, the disease serves as a model for precision medicine, with treatment selection guided by the specific mutation subtype in every patient.
Table of Contents
- Overview
- Cell of Origin
- Molecular Drivers
- Clinical Presentation
- Diagnosis & Pathology
- Risk Stratification
- Treatment: Localized Disease
- Treatment: Metastatic Disease
- Prognosis & Survival
- References
- Connections
- Featured Videos
Overview
Gastrointestinal stromal tumors are soft-tissue sarcomas that arise from the mesenchyme — the connective-tissue layer of the GI wall — rather than from the mucosal epithelium like adenocarcinomas. They account for roughly 80% of all GI mesenchymal tumors and are estimated to occur in 10–15 per million people per year in the United States, representing approximately 5,000–6,000 new diagnoses annually. GISTs can occur anywhere in the GI tract from the esophagus to the rectum, and very rarely in extra-GI sites such as the omentum, mesentery, and retroperitoneum.
The stomach is the most common site, accounting for 50–60% of cases, followed by the small intestine (20–30%), colorectum (5–10%), esophagus (less than 5%), and extra-GI locations (rare). The anatomical location matters clinically: gastric GISTs tend to have a more favorable prognosis than small-intestinal GISTs of equivalent size and mitotic rate, likely reflecting differences in tumor biology and the immunological milieu rather than purely anatomical factors.
Before 2001, advanced GIST carried a median survival of fewer than 18 months and was essentially chemotherapy-resistant — conventional cytotoxic regimens for soft-tissue sarcoma achieved response rates under 5%. The identification of KIT gain-of-function mutations as the dominant oncogenic driver by Hirota et al. in 1998, followed by the landmark clinical trials of imatinib mesylate published in 2001–2002, fundamentally changed the prognosis. Imatinib produced objective tumor responses in over 50% of metastatic GIST patients and disease control in over 80%, extending median survival from under 18 months to over 5 years for patients who respond to first-line therapy.
GIST remains biologically heterogeneous. While most tumors harbor activating mutations in KIT or PDGFRA, a clinically distinct subset — "wildtype" GIST — lacks these mutations and frequently occurs in younger patients, women, and those with germline predisposition syndromes. The management of each molecular subtype has become increasingly individualized as successive generations of kinase inhibitors have entered practice.
Cell of Origin: Interstitial Cells of Cajal
GISTs arise from, or differentiate along the lineage of, the interstitial cells of Cajal (ICC) — a specialized population of mesenchymal cells distributed throughout the GI wall that function as the pacemaker cells of GI smooth muscle. The ICCs generate the slow-wave electrical rhythms that drive coordinated peristalsis and mediate neurotransmission between enteric motor neurons and smooth muscle cells. They form a three-dimensional network within the myenteric plexus and between the circular and longitudinal muscle layers.
The ICC lineage is defined by constitutive expression of three molecular markers that are central to the diagnosis of GIST:
- KIT (CD117) — a receptor tyrosine kinase encoded by the proto-oncogene c-KIT on chromosome 4q12. KIT is expressed on ICCs and is essential for ICC development, proliferation, and function. It is present on over 95% of GISTs and was the first identified marker that distinguished GISTs from other GI mesenchymal tumors such as leiomyoma and schwannoma. KIT expression in GIST is typically strong and diffuse on immunohistochemistry, in contrast to the weak/focal staining seen in some other KIT-expressing tumors.
- DOG1 (TMEM16A, ANO1) — a calcium-activated chloride channel protein encoded on chromosome 11q13. DOG1 (discovered in a gene expression profiling study in 2004, named for "Discovered on GIST-1") is expressed in approximately 95–100% of GISTs including 30–40% of KIT-negative GISTs, making it the most sensitive single marker currently available. DOG1 is highly selective for GIST among GI mesenchymal tumors. Its physiological role on ICCs relates to chloride transport and may contribute to the pacemaker current.
- CD34 — a hematopoietic progenitor and endothelial cell antigen expressed on approximately 60–70% of GISTs, particularly those arising in the stomach. CD34 is less specific than KIT or DOG1 but adds value to the diagnostic panel when the other markers are equivocal.
The evidence that GISTs originate from ICCs or ICC precursors rests on: (1) the shared molecular signature (KIT+, DOG1+, CD34+); (2) ultrastructural similarities between GIST cells and ICCs (dense granules, cell processes, gap junctions); (3) the normal physiological requirement for KIT signaling in ICC development (mice with KIT loss-of-function mutations lack ICCs entirely); and (4) the gain-of-function KIT mutations found in GISTs recapitulate the constitutive KIT activation needed for ICC pacemaker activity. It is likely that KIT-activating mutations in ICC progenitors drive transformation, whereas the tumor cells then retain ICC-lineage markers throughout their development.
Molecular Drivers
GIST is one of the most molecularly well-characterized solid tumors. In most patients, a single driver mutation in one of a small number of genes determines the biological behavior and, critically, the response to specific tyrosine kinase inhibitors. Mutation testing is now considered mandatory before initiating systemic therapy.
KIT Mutations (75–80% of GISTs)
KIT encodes the KIT receptor tyrosine kinase. Normally, KIT requires binding of its ligand stem cell factor (SCF) to dimerize and autophosphorylate, activating downstream signaling pathways including RAS/MAPK, PI3K/AKT, and STAT3/5. In GIST, gain-of-function mutations cause ligand-independent, constitutive KIT activation — the kinase is permanently "on," driving uncontrolled cell proliferation and survival.
- Exon 11 mutations (60–70% of all GISTs) — the most common site, encoding the juxtamembrane domain. Mutations include in-frame deletions (most common), point mutations, and duplications. Exon 11 GISTs have the best response to imatinib 400 mg/day — objective response rates of 65–70% in clinical trials versus 40–50% for other KIT mutations. Deletions involving codons 557–558 within exon 11 are associated with particularly aggressive behavior and higher recurrence risk even after complete resection.
- Exon 9 mutations (10–15%) — encoding the extracellular domain, typically a 6-base-pair duplication (AY502–503dup). More common in small-intestinal GISTs than gastric. Standard imatinib 400 mg/day is less effective; data from randomized trials support imatinib 800 mg/day for this mutation subtype in the metastatic setting, improving response rates from ~17% to ~37%.
- Exon 13 and 17 mutations (rare, ~2% each) — encoding the ATP-binding pocket (exon 13) and the activation loop (exon 17). Generally imatinib-sensitive but require confirmation. Secondary exon 17 mutations are a common mechanism of acquired imatinib resistance.
PDGFRA Mutations (10% of GISTs)
PDGFRA encodes platelet-derived growth factor receptor alpha, a structurally related receptor tyrosine kinase on chromosome 4q12 adjacent to KIT. PDGFRA-mutant GISTs tend to arise preferentially in the stomach, show epithelioid cell morphology, have lower mitotic rates, and behave more indolently than KIT-mutant GISTs of equivalent size.
- PDGFRA D842V (exon 18) — a valine-for-aspartate substitution at codon 842 of the activation loop. Accounts for approximately 65–70% of all PDGFRA mutations and is the most clinically important because it confers near-complete primary resistance to imatinib and sunitinib. Avapritinib, a switch-control kinase inhibitor specifically designed to overcome D842V resistance, received FDA approval in January 2020 for this specific mutation subtype.
- Other PDGFRA mutations (exon 12 and exon 14) — generally imatinib-sensitive.
Wildtype GIST (10–15%)
GISTs lacking KIT or PDGFRA mutations are classified as "wildtype" and comprise a heterogeneous group with distinct clinical and pathological features. They are more common in children, young adults, and women (in whom they may arise in the syndromic setting), and tend to cluster in the stomach.
- SDH-deficient GIST — the most common wildtype subtype. Loss of function of the succinate dehydrogenase (SDH) complex, through mutations in SDHA, SDHB, SDHC, or SDHD, leads to accumulation of succinate and a hypermethylator epigenetic phenotype. SDH-deficient GISTs are KIT- and DOG1-positive but negative for SDHB on IHC (regardless of which SDH subunit is mutated). They frequently arise in the stomach, occur in children and young adults, have an indolent course with frequent lymph node metastases (unusual in KIT/PDGFRA-mutant GIST), and are often refractory to imatinib. SDH deficiency may be germline (Carney-Stratakis syndrome: paraganglioma + GIST) or somatic/epigenetic (Carney triad: GIST + pulmonary chondroma + paraganglioma in young women).
- NF1-associated GIST — patients with neurofibromatosis type 1 have a markedly elevated lifetime risk of developing GIST, typically multiple small-intestinal tumors with low mitotic rates. These tumors are KIT/PDGFRA wildtype and NF1-driven; they are largely imatinib-insensitive.
- BRAF-mutant GIST (V600E and others) — rare (<1% of all GISTs), but clinically important because BRAF V600E mutations render tumors imatinib-resistant. BRAF-mutant GISTs may be sensitive to RAF/MEK inhibitors.
Clinical Presentation
The clinical presentation of GIST is highly variable and depends on tumor size, location, and whether the tumor grows intraluminally (into the GI lumen), exophytically (outward from the GI wall), or endophytically within the wall. Small GISTs — particularly those in the stomach — are often clinically silent and are discovered incidentally on endoscopy performed for unrelated reasons such as evaluation of reflux, Barrett esophagus surveillance, or dyspepsia.
Incidental Discovery: With the widespread use of upper endoscopy and cross-sectional imaging, incidental gastric GISTs smaller than 2 cm are increasingly identified. These are usually submucosal lesions with intact overlying mucosa and smooth contours on endoscopy; the endoscopic appearance alone cannot reliably distinguish GIST from leiomyoma, schwannoma, or lipoma, making endoscopic ultrasound (EUS) and tissue sampling essential for characterization.
Symptomatic Presentations:
- GI bleeding — the most common symptom of clinically presenting GIST. Larger tumors undergo central necrosis as they outgrow their blood supply, creating an ulcer at the mucosa-tumor interface that bleeds into the lumen. Bleeding may be insidious (guaiac-positive stool, iron-deficiency anemia) or acute (hematemesis, melena, hematochezia). GI hemorrhage is particularly characteristic of small-intestinal GISTs.
- Abdominal pain or discomfort — dull, nonspecific pain related to the tumor mass effect or, in larger tumors, stretching of the mesentery or adjacent organs.
- Palpable abdominal mass — large tumors may be palpable on physical examination. GISTs can grow to impressive sizes (10–30 cm) before causing symptoms, particularly when growing exophytically away from the lumen.
- Obstruction — duodenal or pyloric GISTs may cause gastric outlet obstruction; small-intestinal GISTs may cause partial or complete obstruction. Esophageal GISTs cause progressive dysphagia.
- Perforation — rare but life-threatening; may be the presenting event for large, necrotic tumors.
Metastatic patterns: GIST spreads hematogenously, most commonly to the liver (in 50–65% of metastatic cases) and peritoneum. Lymph node metastases are rare in KIT/PDGFRA-mutant GIST (unlike most carcinomas), which has important implications for surgical technique — lymph node dissection adds morbidity without benefit. Lung and bone metastases occur but are uncommon.
Diagnosis & Pathology
The diagnosis of GIST requires tissue biopsy with histological examination and immunohistochemical staining. Imaging is essential for staging and surgical planning but cannot provide a tissue diagnosis.
Imaging
Contrast-enhanced CT of the chest, abdomen, and pelvis is the standard imaging modality for staging. GISTs typically appear as well-defined, hypervascular solid masses arising from the GI wall with avid arterial-phase enhancement. Larger tumors commonly show central necrosis, cystic degeneration, and heterogeneous attenuation. The hypervascular blush distinguishes GIST from most GI carcinomas (which are hypovascular). Metastatic deposits in the liver are also typically hypervascular. PET-CT adds metabolic assessment and is valuable for early response monitoring — imatinib-responding tumors often show dramatic FDG-PET responses within days of treatment initiation, long before CT shows a change in tumor size.
Endoscopy and Endoscopic Ultrasound (EUS): Upper endoscopy visualizes the overlying mucosa and can identify the submucosal origin and surface ulceration of GIST. EUS characterizes the wall layer of origin (fourth layer — muscularis propria — for most GISTs, distinguishing them from submucosal lipomas which arise in the third layer), internal echogenicity, and size. EUS-guided fine-needle aspiration (EUS-FNA) or biopsy allows tissue sampling for histology and IHC, particularly for gastric GISTs when percutaneous CT-guided biopsy would risk peritoneal seeding.
Biopsy and Histology
GISTs most commonly show one of three histological patterns:
- Spindle cell type (70%) — the most common; cells are elongated with eosinophilic cytoplasm, arranged in fascicles. Mitotic figures are key to risk assessment.
- Epithelioid type (20%) — round to polygonal cells with abundant pale or clear cytoplasm; more commonly associated with PDGFRA mutations, particularly the D842V variant.
- Mixed spindle and epithelioid (10%).
Immunohistochemistry (IHC)
IHC is the cornerstone of GIST diagnosis. The standard panel includes:
- CD117 (KIT) — positive in ~95% of GISTs; typically diffuse cytoplasmic staining with a membranous pattern. The rare KIT-negative GISTs (approximately 5%) are mostly PDGFRA-mutant epithelioid tumors.
- DOG1 — positive in ~95–100% of GISTs, including many KIT-negative cases. When combined with CD117, sensitivity approaches 99%. DOG1 has essentially replaced CD34 as the second-line marker in most expert centers.
- CD34 — positive in 60–70%, supporting GIST diagnosis in the right context.
- SDHB — loss of SDHB staining (regardless of which SDH subunit is mutated) identifies SDH-deficient wildtype GIST; should be tested in all KIT/PDGFRA wildtype cases and in pediatric/young adult GISTs.
- Ki-67 (MIB-1) — proliferative index; higher values correlate with more aggressive behavior but the mitotic count per standardized area (50 HPF or 5 mm²) remains the primary measure.
Molecular Testing
Mutation analysis of KIT (exons 9, 11, 13, 17) and PDGFRA (exons 12, 14, 18) is now standard of care for all GISTs requiring systemic therapy. The specific mutation determines the choice and dose of tyrosine kinase inhibitor. For KIT/PDGFRA wildtype cases, extended panel testing including SDHA/B/C/D, NF1, BRAF, and RAS should be performed to identify actionable alterations and inform prognosis.
Risk Stratification
Not all GISTs behave the same way. Small GISTs — particularly those smaller than 2 cm in the stomach — almost never metastasize, while large GISTs with high mitotic activity carry a high risk of recurrence even after complete surgical resection. Accurately stratifying risk is essential for deciding who needs adjuvant systemic therapy after curative-intent surgery.
The NIH/Fletcher consensus criteria (2002), subsequently modified by Miettinen and Lasota (2006) incorporating anatomical site, provide the most widely used stratification system. Risk is classified based on two primary variables:
- Tumor size — measured as the largest diameter on pathological specimen.
- Mitotic rate — expressed as the number of mitoses per 50 high-power fields (HPF) or, in updated guidelines, per 5 mm² of tumor area; threshold is 5 per 50 HPF.
Risk categories (gastric GIST):
- Very low risk — size <2 cm AND mitotic rate ≤5/50 HPF. No adjuvant therapy recommended. Annual surveillance imaging for 5 years is reasonable for tumors close to the threshold.
- Low risk — size 2–5 cm AND mitotic rate ≤5/50 HPF. Very low metastatic rate (<4% at 5 years in gastric primaries).
- Intermediate risk — size <5 cm AND mitotic rate 6–10/50 HPF, OR size 5–10 cm AND mitotic rate ≤5/50 HPF. Metastatic rate approximately 10–25%.
- High risk — any of: size >10 cm (any mitotic rate), mitotic rate >10/50 HPF (any size), size >5 cm with mitotic rate >5/50 HPF, or tumor rupture. Metastatic rate 50–90%.
Site effect: Non-gastric location independently worsens prognosis at any given size/mitotic combination. A 5 cm small-intestinal GIST with mitotic rate <5/50 HPF carries a much higher metastatic risk (~24%) than a comparable gastric GIST (~4%). This anatomical modifier is incorporated into the Miettinen/Lasota contour maps, which provide more granular risk estimates than the four-category NIH system.
Tumor rupture is an independent high-risk factor regardless of size or mitotic rate. Rupture — whether spontaneous, at surgery, or from iatrogenic perforation during biopsy — spills tumor cells into the peritoneal cavity and dramatically increases the risk of peritoneal recurrence. Some guidelines consider ruptured GIST equivalent to metastatic disease in terms of the indication for indefinite imatinib therapy rather than adjuvant-duration treatment.
Exon 11 deletion type: Within high-risk GISTs, the specific KIT exon 11 mutation subtype provides additional prognostic information. Deletions encompassing codons 557–558 are associated with particularly high recurrence risk and may warrant extended or indefinite adjuvant imatinib duration in future clinical trials.
Treatment: Localized Disease
Surgery is the only potentially curative treatment for localized GIST, and the operative principles differ in important ways from the management of carcinomas at the same anatomical sites.
Surgery
Goal: Complete macroscopic resection with negative histological margins (R0). The target negative margin width is debated; because GISTs grow by expansion rather than infiltration, close margins (<1 cm) may still be oncologically adequate provided there is no gross residual tumor and no tumor rupture. Pseudocapsule preservation — avoiding rupture of the tumor's fibrous capsule during manipulation — is more important than achieving a wide soft-tissue margin.
No lymph node dissection: Unlike carcinomas arising at the same anatomical sites (gastric adenocarcinoma, colorectal cancer), KIT/PDGFRA-mutant GISTs virtually never metastasize to regional lymph nodes. Systematic lymphadenectomy adds morbidity without oncological benefit and is not performed. This is one of the most operationally important distinctions when surgeons accustomed to GI oncology operate on GISTs.
Laparoscopic resection is appropriate for gastric and small-intestinal GISTs under 5 cm in accessible locations; outcomes equivalent to open surgery provided the tumor is not ruptured. Tumors larger than 5 cm at challenging locations (gastroesophageal junction, duodenum, rectum) often require open surgery or multidisciplinary planning.
Neoadjuvant Imatinib
For tumors that are technically resectable but where surgery would require extensive organ sacrifice (e.g., a large duodenal GIST requiring pancreaticoduodenectomy, a rectal GIST requiring abdominoperineal resection), pre-operative imatinib therapy can shrink the tumor and allow a less morbid operation. Neoadjuvant imatinib is administered for 6–12 months with response assessment every 2–3 months by CT and/or PET; surgery is performed when maximal response has been achieved. Mutation testing before starting neoadjuvant therapy is essential — PDGFRA D842V and NF1-associated GISTs will not respond and should proceed directly to surgery.
Adjuvant Imatinib
The American College of Surgeons Oncology Group (ACOSOG) Z9001 trial (DeMatteo et al., Lancet 2009) demonstrated that one year of adjuvant imatinib 400 mg/day after complete resection of KIT-positive GIST ≥3 cm improved recurrence-free survival compared to placebo (98% vs. 83% at 1 year). The subsequent Scandinavian Sarcoma Group/Arbeitsgemeinschaft Internistische Onkologie (SSG XVIII) trial (Joensuu et al., JAMA 2012) showed that 3 years of adjuvant imatinib was superior to 1 year for high-risk patients, improving 5-year recurrence-free survival (65.6% vs. 47.9%) and overall survival (92% vs. 81.7%).
Based on these trials, 3 years of adjuvant imatinib is the current standard of care for high-risk GIST (high-risk per NIH criteria). Adjuvant therapy is not recommended for very-low or low-risk disease; evidence for intermediate-risk patients is less clear. An ongoing clinical question is whether 3 years is sufficient for very high-risk patients (tumor rupture, very large size with high mitotic rate) — some centers advocate 5 years or indefinite therapy in these cases.
Treatment: Metastatic and Unresectable Disease
The systemic treatment of metastatic GIST follows a line-therapy sequence of increasingly selective tyrosine kinase inhibitors, guided at each step by the mutation profile and the mechanism of resistance to prior therapy.
First-Line: Imatinib Mesylate (Gleevec)
Imatinib, an orally bioavailable ABL/KIT/PDGFRA kinase inhibitor, is the cornerstone of first-line treatment for KIT-mutant metastatic GIST. The pivotal Phase II trial reported by Demetri et al. in the New England Journal of Medicine (2002) enrolled 147 patients with advanced GIST and achieved a partial response rate of 54% and disease control (partial response + stable disease) in 84% of patients — a result unprecedented in soft-tissue sarcoma. Median survival in this trial exceeded 57 months, compared to fewer than 18 months with conventional chemotherapy.
- Standard dose: 400 mg/day for KIT exon 11 mutations (best response) and PDGFRA non-D842V mutations.
- Dose escalation to 800 mg/day: Randomized data support 800 mg/day as initial dose for KIT exon 9 mutations in the metastatic setting; response rates approximately double those seen with 400 mg/day for this genotype.
- PDGFRA D842V: Imatinib produces response rates under 10% — this mutation should be specifically identified before starting therapy, as avapritinib is the appropriate first-line agent.
- Wildtype GIST: SDH-deficient and NF1-associated GISTs show minimal benefit from imatinib; disease control may still occur in some patients.
Common imatinib toxicities include edema (periorbital, lower extremity), nausea, diarrhea, muscle cramps, and fatigue. Most are manageable and dose modification is rarely required. Serious toxicities (hepatotoxicity, severe myelosuppression) are uncommon.
Second-Line: Sunitinib
Sunitinib (Sutent), a multi-targeted inhibitor of KIT, PDGFR, VEGFR, FLT3, and RET, is the standard second-line treatment after imatinib failure or intolerance. A Phase III trial demonstrated that sunitinib significantly improved time to tumor progression (27.3 vs. 6.4 weeks) and overall survival compared to placebo following imatinib failure. Sunitinib is particularly active against secondary KIT exon 13 and 14 mutations, which are among the most common mechanisms of acquired imatinib resistance. It is less active against secondary exon 17/18 mutations. The standard dose is 50 mg/day for 4 weeks on, 2 weeks off, though continuous 37.5 mg/day dosing is increasingly used to reduce toxicity.
Third-Line: Regorafenib
Regorafenib (Stivarga), a broad-spectrum kinase inhibitor targeting KIT, PDGFR, VEGFR, RAF, and other kinases, demonstrated improved progression-free survival versus placebo (4.8 vs. 0.9 months, HR 0.27) in the GRID trial (Demetri et al., Lancet Oncology 2013) after failure of both imatinib and sunitinib, establishing it as the third-line standard. Significant toxicities include hand-foot skin reaction, hypertension, and fatigue. The standard dose is 160 mg/day for 3 weeks on, 1 week off.
Fourth-Line: Ripretinib
Ripretinib (Qinlock) is a switch-control kinase inhibitor that binds both the active and inactive conformations of KIT and PDGFRA, providing activity against a broad spectrum of primary and secondary resistance mutations. The INVICTUS trial (Bauer et al., Lancet Oncology 2020) randomized patients who had received at least three prior lines of therapy to ripretinib 150 mg/day vs. placebo, demonstrating improved median PFS (6.3 vs. 1.0 months, HR 0.36) and OS (15.1 vs. 6.6 months). Ripretinib was approved by the FDA in May 2020 as the fourth-line standard of care.
PDGFRA D842V-Specific: Avapritinib
Avapritinib (Ayvakit) was specifically designed to overcome the imatinib-resistance conferred by the PDGFRA D842V mutation. The NAVIGATOR trial (Heinrich et al., Cancer Discovery 2020) demonstrated an objective response rate of 88% (including 9% complete responses) in patients with PDGFRA D842V-mutant GIST — responses not achievable with any prior agent. Avapritinib was approved by the FDA in January 2020 as the first-ever treatment specifically for PDGFRA D842V GIST and should be used as first-line therapy in this mutation subtype rather than imatinib. At higher doses, avapritinib can cause intracranial bleeding in a subset of patients, requiring careful monitoring; the approved dose for GIST (300 mg/day) has an acceptable toxicity profile.
Resistance Mechanisms
Secondary resistance to imatinib develops in most patients over time through acquisition of additional mutations in the KIT kinase domain that sterically or allosterically interfere with imatinib binding. The most common secondary mutations occur in the ATP-binding pocket (exons 13–14) or the activation loop (exons 17–18). Because multiple resistant clones with different secondary mutations often coexist at the time of progression — clonal heterogeneity — no single second-line agent covers all possible resistance mutations, which explains the limited duration of benefit seen with sequential therapy.
Prognosis & Survival
The prognosis of GIST has been transformed by targeted therapy and is now strongly dependent on three factors: mutation status, risk group at presentation, and line of therapy at the time disease becomes incurable. The contrast between the pre-imatinib and post-imatinib eras represents one of the most striking improvements in the history of oncology for any solid tumor.
Localized GIST after surgery:
- Very low risk: 5-year overall survival exceeds 95%; recurrence risk under 5%. Surveillance alone after resection.
- Low risk: 5-year OS 85–90%; recurrence risk 4–8%.
- Intermediate risk: 5-year OS 70–80% with modern management. Adjuvant imatinib benefit less well established in this group.
- High risk (3 years of adjuvant imatinib): 5-year OS 90%, recurrence-free survival ~65% at 5 years (SSG XVIII trial with 3-year adjuvant regimen). The 3-year adjuvant strategy has dramatically shifted outcomes in this group compared to surgery alone (historical 5-year OS approximately 50%).
Metastatic GIST:
- Historical (pre-imatinib): Median survival 18–24 months; no effective systemic therapy; conventional chemotherapy response rates under 5%.
- First-line imatinib (KIT exon 11): Median progression-free survival 20–24 months; median overall survival 5+ years in responders. Approximately 10–15% of patients treated with first-line imatinib achieve a complete radiological response and may enjoy prolonged disease control exceeding 10 years.
- First-line imatinib (KIT exon 9): Median PFS 13 months at 400 mg/day; improved to approximately 19 months with 800 mg/day.
- PDGFRA D842V (avapritinib first-line): Response rate 88%, median duration of response not reached at data cutoff in NAVIGATOR trial — this mutation subtype, previously carrying a poor prognosis on imatinib, now has an exceptional outlook.
- Sequential therapy across four lines: Median OS from metastatic diagnosis with optimal sequential imatinib → sunitinib → regorafenib → ripretinib approaches 5–7 years in patients who remain on therapy.
Prognostic factors in metastatic disease: Initial response to imatinib (partial response vs. stable disease), tumor burden, number of metastatic sites, and performance status are the strongest predictors of long-term outcome. Patients who achieve a complete metabolic response on PET after imatinib, though rare, can have prolonged disease control exceeding a decade. Peritoneal dissemination carries a somewhat worse prognosis than liver-only metastases.
Wildtype GIST prognosis: SDH-deficient GISTs follow an indolent but persistently progressive course; long-term survivors are common even in metastatic disease, with some patients living 10–15 years after metastatic diagnosis despite imatinib resistance. NF1-associated multifocal small-intestinal GISTs are generally low-grade, rarely fatal from GIST itself, and managed expectantly unless symptomatic.
References
- Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002 Aug 15;347(7):472–80 — Search PubMed. DOI: 10.1056/NEJMoa020461
- DeMatteo RP, Ballman KV, Antonescu CR, et al.; ACOSOG Z9001 investigators. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour. Lancet. 2009 Mar 28;373(9669):1097–104. PMID: 18235122. DOI: 10.1016/S0140-6736(09)60500-6
- Joensuu H, Eriksson M, Sundby Hall K, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor. JAMA. 2012 Jun 27;307(12):1265–72 — Search PubMed. DOI: 10.1001/jama.2012.347
- Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol. 2002 May;33(5):459–65 — Search PubMed. DOI: 10.1053/hupa.2002.123545
- Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998 Jan 23;279(5350):577–80 — Search PubMed. DOI: 10.1126/science.279.5350.577
- Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003 Jan 31;299(5607):708–10 — Search PubMed. DOI: 10.1126/science.1079666
- Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors. J Clin Oncol. 2008 Feb 1;26(4):626–32 — Search PubMed. DOI: 10.1200/JCO.2007.13.4452
- Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID). Lancet. 2013 Jan 26;381(9863):295–302. PMID: 23177515. DOI: 10.1016/S0140-6736(12)61857-1
- Bauer S, George S, von Mehren M, et al. Early and sustained efficacy with avapritinib in relapsed/refractory PDGFRA D842V-mutant gastrointestinal stromal tumors. Lancet Oncol. 2020 Feb;21(2):263–74 — Search PubMed. DOI: 10.1016/S1470-2045(19)30816-4
- Heinrich MC, Jones RL, von Mehren M, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour. Cancer Discov. 2020 Jul;10(7):1034–49. PMID: 32511981. DOI: 10.1158/2159-8290.CD-20-0069
- Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006 May;23(2):70–83 — Search PubMed. DOI: 10.1053/j.semdp.2006.09.001
- Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011 Dec 22;11(12):865–78. PMID: 22089421. DOI: 10.1038/nrc3143
Search additional GIST literature: PubMed — Gastrointestinal Stromal Tumor
Connections
- Oncology
- Colorectal Cancer
- Stomach Cancer
- Cholangiocarcinoma
- Pancreatic Cancer
- SIBO
- Mesothelioma
- Cancer Overview
- Gastroenterology
- Lab Tests