Neuroblastoma
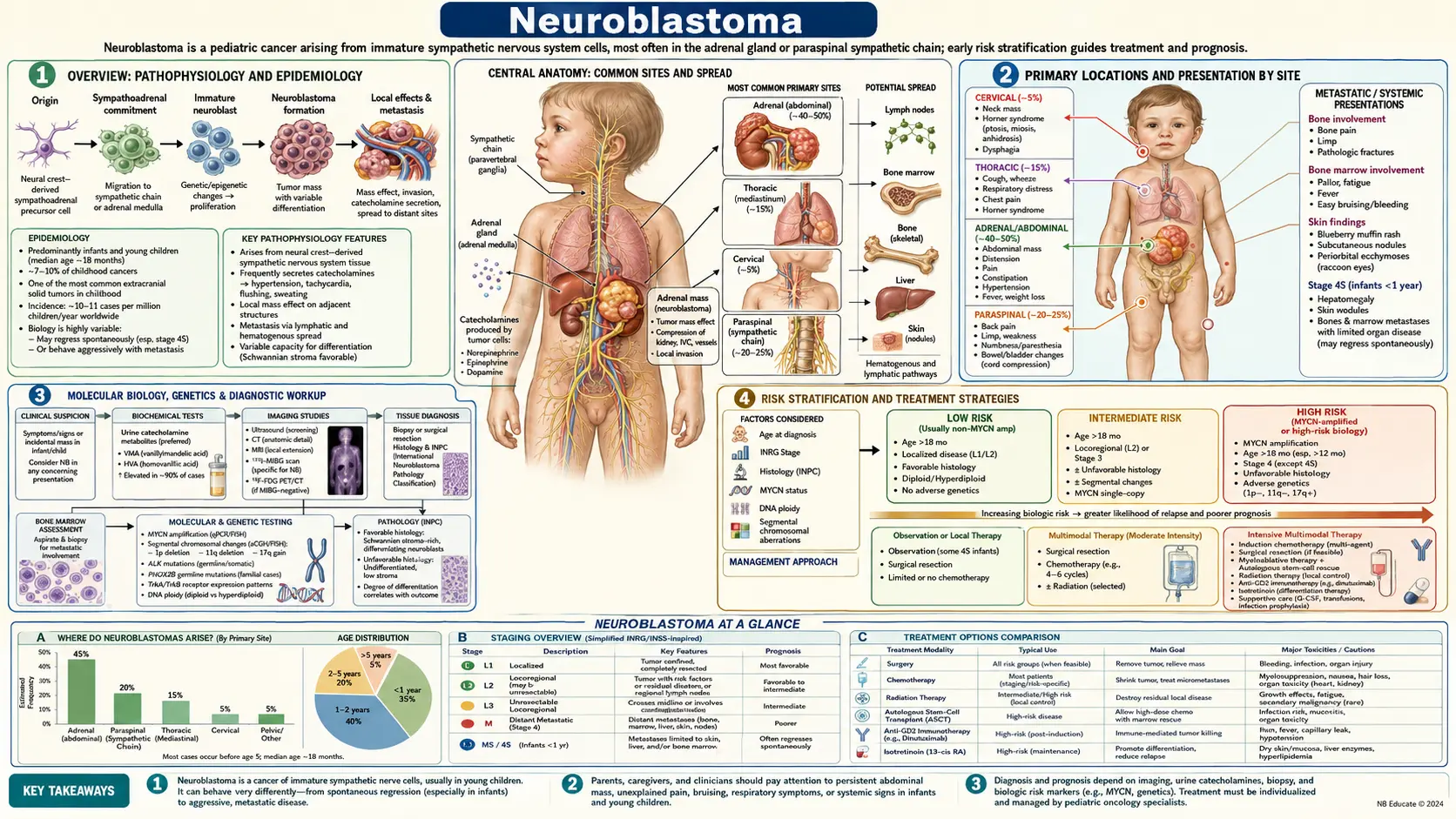
Table of Contents
- Overview
- Primary Locations and Presentation by Site
- Molecular Biology and Genetics
- Risk Stratification
- Diagnosis and Staging
- Treatment by Risk Group
- Prognosis
- Emerging Therapies and Research
- Key Research Papers
- Featured Videos
- Connections
Overview
Neuroblastoma is the most common extracranial solid tumor of childhood and the most frequently diagnosed cancer in infancy. In the United States, approximately 700 to 800 new cases are diagnosed each year, making it the third most common pediatric cancer after leukemia and brain tumors. The peak age at diagnosis is around 2 years, and roughly 90% of cases are diagnosed before age 10.
The tumor arises from neural crest cells — embryonic progenitors that give rise to the sympathetic nervous system, specifically from the sympathoadrenal lineage of chromaffin cells and sympathetic neuroblasts. Because neural crest cells migrate throughout the body during fetal development, neuroblastoma can arise anywhere along the sympathetic chain, from the neck to the pelvis, with the adrenal medulla being the single most common primary site.
Neuroblastoma is responsible for approximately 15% of all pediatric cancer deaths despite representing only 7 to 8% of childhood cancers — a stark disparity that reflects the aggressive behavior of high-risk disease. What makes neuroblastoma biologically unique among childhood cancers is its remarkably wide clinical spectrum. At one extreme, infants with localized or metastatic-special (MS) disease may undergo spontaneous complete regression without any treatment. At the other extreme, older children with high-risk metastatic neuroblastoma face a relentless disease course with historically fewer than one in three patients achieving long-term survival, though outcomes have improved significantly with modern immunotherapy protocols.
This biological heterogeneity — from self-resolving lesion to one of the deadliest childhood cancers — has driven decades of research into the molecular drivers that distinguish favorable from unfavorable disease, and has made neuroblastoma a model for precision oncology in pediatrics.
Primary Locations and Presentation by Site
The location of the primary tumor determines the initial clinical presentation, and recognizing these site-specific patterns is essential for early diagnosis.
Primary Site Distribution
- Adrenal medulla: Most common primary site, accounting for approximately 40% of all neuroblastomas. These tumors arise directly from the adrenal chromaffin cells.
- Paraspinal retroperitoneum: About 25% of cases arise from sympathetic ganglia along the retroperitoneal chain, adjacent to the vertebral column.
- Posterior mediastinum: Approximately 15% arise from paraspinal ganglia in the chest, often discovered incidentally on chest X-ray.
- Cervical: About 3 to 5% arise from the superior cervical sympathetic ganglion or adjacent ganglia in the neck.
- Pelvic: About 3 to 5% arise from the organ of Zuckerkandl or pelvic sympathetic chain.
Abdominal Presentation (Most Common)
Abdominal neuroblastoma typically presents as a firm, fixed, hard, irregular abdominal mass that frequently crosses the midline. This distinguishes it from Wilms tumor (nephroblastoma), which is characteristically smooth, mobile, and confined to one side. Parents often notice the mass incidentally while bathing the child. Associated symptoms may include abdominal distension, decreased appetite, and irritability. Hypertension can occur due to catecholamine secretion from the tumor or compression of the renal vasculature.
Orbital Metastases — "Raccoon Eyes"
One of the most distinctive signs in metastatic neuroblastoma is periorbital ecchymosis — bilateral bruising around the eyes giving the appearance of "raccoon eyes" — combined with proptosis (forward protrusion of the eyeball). This results from metastatic tumor deposits in the orbital bones. Parents frequently bring children to the emergency department believing the child has been physically abused, making recognition of this sign critical. The bilateral and painless nature, absence of trauma history, and associated systemic findings (pallor, irritability, bone pain) should prompt immediate evaluation for neuroblastoma.
Cervical Presentation — Horner Syndrome
Cervical neuroblastoma involving the superior cervical sympathetic ganglion causes Horner syndrome: ipsilateral ptosis (drooping upper eyelid), miosis (constricted pupil), and anhidrosis (absence of sweating on that side of the face). In infants, heterochromia iridis (different colored irises) may also be present if the sympathetic injury occurred during the critical period of iris pigmentation development. A neck mass may or may not be palpable.
Thoracic Presentation
Posterior mediastinal neuroblastoma can cause respiratory distress from airway compression, superior vena cava (SVC) syndrome with facial swelling and venous distension, dysphagia, and recurrent respiratory infections. These tumors are often discovered incidentally on chest X-ray obtained for another reason, appearing as a posterior mediastinal mass in the paravertebral sulcus.
Paraspinal "Dumbbell" Tumors
Paraspinal neuroblastomas are particularly dangerous because they can extend through the neural foramina into the spinal canal — creating a "dumbbell" configuration with both an extraspinal and intraspinal component. Epidural extension can compress the spinal cord, causing back pain, weakness, gait disturbance, and if not treated urgently, permanent paraplegia. Any child with a paraspinal mass and new neurological symptoms requires emergent MRI of the spine.
Paraneoplastic — Opsoclonus-Myoclonus-Ataxia (OMA) Syndrome
Approximately 2 to 4% of neuroblastoma patients develop opsoclonus-myoclonus-ataxia (OMA) syndrome, sometimes called "dancing eyes-dancing feet" syndrome. The clinical triad consists of:
- Opsoclonus: Chaotic, multidirectional involuntary eye movements without saccadic intervals
- Myoclonus: Brief, involuntary muscle jerks, particularly of the trunk and limbs
- Cerebellar ataxia: Unsteady gait, clumsiness, truncal instability
OMA is immune-mediated — the body produces antibodies against neural antigens expressed by the tumor, which then cross-react with cerebellar neurons. Paradoxically, OMA is more commonly associated with localized, low-stage disease and carries a better tumor prognosis. However, the neurological consequences are severe: 50 to 70% of survivors have long-term neurodevelopmental sequelae including cognitive impairment, language delay, and behavioral problems, even after the tumor is treated.
Systemic Symptoms
Systemic features include fever, weight loss, irritability, and pallor from bone marrow involvement. Watery diarrhea can occur in vasoactive intestinal peptide (VIP)-secreting tumors. Hypertension affects a subset of patients from catecholamine secretion. Bone pain and a limp are common presenting complaints in children with skeletal metastases.
Molecular Biology and Genetics
Neuroblastoma has been intensively studied at the molecular level, and several genomic features have direct clinical significance in determining prognosis and guiding therapy.
MYCN Amplification
Amplification of the MYCN oncogene — present in approximately 25% of primary neuroblastomas — is the single most important independent poor prognostic marker. MYCN encodes a transcription factor that drives cell proliferation, suppresses differentiation, and promotes tumor progression. MYCN-amplified tumors are biologically aggressive, associated with rapid clinical progression, and found disproportionately in high-stage disease. MYCN amplification is defined as greater than 10 copies of the MYCN locus (greater than 4-fold increase) relative to ploidy. Even in patients with otherwise favorable features (low stage, young age), MYCN amplification confers high-risk designation and mandates intensive therapy.
ALK Mutations and Amplification
Activating point mutations or amplification of the anaplastic lymphoma kinase (ALK) gene are found in approximately 10% of sporadic neuroblastomas and up to 50% of familial neuroblastoma cases. ALK is a receptor tyrosine kinase, and neuroblastoma-associated mutations (most commonly F1174L, R1275Q, and F1245C) constitutively activate downstream proliferative signaling. ALK is a validated therapeutic target: the ALK/ROS1 inhibitor crizotinib and the next-generation lorlatinib (which penetrates the CNS) are being evaluated in clinical trials for ALK-mutated neuroblastoma.
PHOX2B Mutations
Germline mutations in PHOX2B (paired-like homeobox 2B), a transcription factor essential for sympathetic nervous system development, cause familial neuroblastoma. PHOX2B mutations also underlie congenital central hypoventilation syndrome (Ondine's curse). Children with PHOX2B mutations have a significantly elevated lifetime risk of developing neuroblastoma and require surveillance.
Segmental Chromosomal Aberrations
Segmental chromosomal aberrations (SCAs) are partial gains or losses of chromosomal regions and carry independent prognostic significance:
- 1p deletion: Loss of the short arm of chromosome 1 is found in 25 to 35% of neuroblastomas. It is associated with MYCN amplification and unfavorable prognosis. The 1p36 region harbors putative tumor suppressor genes.
- 11q deletion: Loss of 11q material is found in approximately 35% of neuroblastomas — notably often in MYCN non-amplified tumors — and is associated with unfavorable prognosis. Patients with 11q deletion but without MYCN amplification constitute a distinct high-risk subgroup.
- 17q gain: Gain of the long arm of chromosome 17 is the most frequent chromosomal abnormality in neuroblastoma (present in ~50%), associated with aggressive disease and poor prognosis.
DNA Ploidy
The DNA index (DI) — the ratio of the tumor's DNA content to a normal diploid cell — has prognostic significance primarily in infants. Hyperdiploid tumors (DI greater than 1.0, typically triploid) in infants with low-stage disease are associated with favorable outcomes and chemosensitivity. Diploid tumors (DI = 1.0) in the same age group tend to behave more aggressively. In older children with high-stage disease, ploidy loses its independent prognostic value.
ATRX Mutations and Telomere Lengthening
Mutations in ATRX (a chromatin remodeling gene) occur in approximately 17% of neuroblastomas and are strongly associated with older age at diagnosis, absence of MYCN amplification, and a distinct pattern of late relapse. ATRX-mutated tumors activate alternative lengthening of telomeres (ALT), conferring cellular immortality through a recombination-based mechanism rather than telomerase. Telomere lengthening mechanisms — whether through ATRX loss/ALT or TERT (telomerase reverse transcriptase) genomic rearrangements — define a high-risk, late-relapsing genomic subgroup that requires specialized monitoring.
Risk Stratification
Accurate risk stratification is the cornerstone of neuroblastoma management — it determines the intensity of treatment and has allowed thousands of children with low-risk disease to be spared the toxicity of chemotherapy, while concentrating intensive multimodal therapy on those with high-risk disease.
Image-Defined Risk Factors (IDRFs)
The International Neuroblastoma Risk Group (INRG) staging system, adopted in 2009, uses preoperative imaging to define image-defined risk factors (IDRFs) — features that predict surgical complexity and risk of major complications. IDRFs include tumor encasement of major blood vessels, infiltration of adjacent vital organs, and extension into critical anatomical compartments. The presence or absence of IDRFs at diagnosis determines the INRG surgical stage and guides upfront surgical decision-making: surgeons avoid aggressive resection when IDRFs are present, instead using chemotherapy to reduce tumor volume before attempting resection.
INRG Staging
- L1: Localized tumor confined to one body compartment, no IDRFs present. Amenable to complete surgical resection upfront.
- L2: Localized tumor with one or more IDRFs present. Surgery deferred until after chemotherapy reduces risk.
- M: Distant metastatic disease (beyond regional lymph nodes). Corresponds to high-risk designation in children older than 18 months.
- MS: Metastatic-special stage. Applies only to children younger than 18 months. Metastases are limited to skin, liver, and/or bone marrow (without cortical bone involvement). Associated with high rates of spontaneous regression.
INRG Risk Groups
The INRG Task Force defined four risk groups based on pretreatment factors: stage, age at diagnosis, histological grade, MYCN amplification status, DNA ploidy, and segmental chromosomal aberrations:
- Very Low Risk: Expected event-free survival (EFS) greater than 85%. Minimal or no therapy required. Some infants with small adrenal masses can be observed without biopsy.
- Low Risk: Expected EFS 75 to 85%. Surgery alone or with brief, moderate-intensity chemotherapy.
- Intermediate Risk: Expected EFS 50 to 75%. Surgery combined with moderate-intensity chemotherapy.
- High Risk: Expected EFS less than 50% with conventional treatment. Approximately 40% of all neuroblastoma patients. Requires intensive multimodal therapy including induction chemotherapy, surgery, high-dose consolidation with stem cell rescue, radiation, and immunotherapy maintenance.
High-Risk Designation Criteria
A patient is classified as high-risk if they have: Stage M disease and are older than 18 months at diagnosis; or any stage disease with MYCN amplification; or Stage L2 disease age 18 months or older with unfavorable histology and segmental chromosomal aberrations. Children younger than 18 months with Stage M disease but favorable biology may be reclassified to intermediate risk after biopsy results.
Diagnosis and Staging
The diagnosis of neuroblastoma requires a combination of biochemical markers, imaging studies, and tissue biopsy for definitive confirmation and molecular profiling.
Urine Catecholamine Metabolites
Because neuroblastoma arises from catecholamine-producing sympathetic tissue, the tumor secretes dopamine, norepinephrine, and epinephrine — which are metabolized to vanillylmandelic acid (VMA) and homovanillic acid (HVA) and excreted in the urine. Elevated 24-hour urine or spot urine VMA and HVA (corrected for creatinine) are found in over 90% of neuroblastoma patients and are highly sensitive and specific diagnostic markers. Serum and urine catecholamine measurement is therefore a first-line biochemical test whenever neuroblastoma is suspected.
Serum Tumor Markers
- Lactate dehydrogenase (LDH): Elevated in high tumor burden; an independent poor prognostic marker in several risk stratification schemas.
- Ferritin: Elevated serum ferritin correlates with high-risk disease and poor outcome; may reflect tumor-derived ferritin secretion.
- Neuron-specific enolase (NSE): An isoenzyme of enolase expressed in neurons and neuroendocrine cells; elevated in many neuroblastoma patients and used for monitoring response, though less specific than catecholamines.
MIBG Scintigraphy
Meta-iodobenzylguanidine (MIBG) is a norepinephrine analog that is taken up by cells with active norepinephrine transporters — including neuroblastoma cells. Radiolabeled MIBG scintigraphy is MIBG-avid in approximately 90% of neuroblastomas and provides whole-body staging information, identifying bone and bone marrow metastases, primary tumor extent, and treatment response. I-123 MIBG is preferred over I-131 MIBG for diagnostic imaging because of superior image quality and lower radiation dose. For the approximately 10% of tumors that are MIBG non-avid, PET-CT using F-18 DOPA or F-18 FDG is used instead.
Cross-Sectional Imaging
CT of the chest, abdomen, and pelvis with contrast provides detailed anatomical characterization of the primary tumor, delineates its relationship to adjacent structures and major vessels, and identifies IDRFs critical for surgical planning. MRI is preferred for paraspinal tumors to assess intraspinal extension and potential cord compression, and for evaluating orbital or intracranial involvement. Brain MRI is not routine but is obtained when neurological symptoms suggest CNS involvement.
Bone Marrow Biopsy
Bilateral posterior iliac crest bone marrow biopsies (core biopsies, not aspirates alone) are required for staging in all patients except those with clearly localized, very-low-risk disease. Bone marrow involvement is found in approximately 70% of high-risk patients at diagnosis and is a component of Stage M disease definition.
Tissue Biopsy and Molecular Profiling
A diagnostic tissue biopsy is required for definitive histological diagnosis and comprehensive molecular profiling including MYCN copy number, ALK mutation status, DNA index (ploidy), segmental chromosomal aberrations by comparative genomic hybridization or SNP array, and histological classification by the International Neuroblastoma Pathology Classification (INPC — Shimada system). The INPC classifies tumors as favorable or unfavorable histology based on degree of neuroblastic differentiation, mitosis-karyorrhexis index (MKI), and patient age. Favorable histology tumors show Schwannian stromal development and low MKI; unfavorable histology tumors are undifferentiated or poorly differentiated with high MKI.
Treatment by Risk Group
Treatment intensity in neuroblastoma is precisely calibrated to risk group, reflecting the goal of curing children with the minimum necessary therapy to avoid long-term toxicity, while deploying the full arsenal of available treatments for high-risk disease.
Very Low and Low Risk
Many infants with small, asymptomatic adrenal masses (detected incidentally on prenatal ultrasound or postnatal imaging) can be managed with careful observation alone — without biopsy or treatment — because a significant proportion spontaneously regress. For symptomatic or growing low-risk tumors, surgical resection alone is curative in most cases. The Children's Oncology Group (COG) established that the majority of low-risk patients require minimal or no chemotherapy, sparing them from treatment toxicity. For infants with Stage MS disease and massive hepatomegaly causing respiratory compromise, low-dose chemotherapy (vincristine, cyclophosphamide) effectively reduces hepatic tumor burden while allowing spontaneous regression to proceed.
Intermediate Risk
Intermediate-risk patients are treated with surgery combined with moderate-intensity chemotherapy, typically 4 to 8 cycles of a carboplatin/etoposide/cyclophosphamide/doxorubicin-based regimen. The COG ANBL0531 trial demonstrated that risk-adapted chemotherapy reduction is feasible in many intermediate-risk patients without compromising survival. Five-year overall survival exceeds 90% in this group.
High Risk — Induction Phase
High-risk neuroblastoma treatment begins with intensive induction chemotherapy aimed at achieving maximum tumor reduction before local control measures. The COG standard induction regimen consists of 6 cycles of multi-agent chemotherapy alternating between:
- Cycles 1, 3, 5: Cyclophosphamide, topotecan, vincristine
- Cycles 2, 4, 6: Carboplatin, etoposide, doxorubicin
Response is assessed by MIBG scintigraphy and cross-sectional imaging. The goal is to achieve at least a partial response before proceeding to surgical resection.
High Risk — Surgery and Local Control
After induction chemotherapy, surgical resection of the primary tumor is performed with the aim of achieving a complete or near-complete resection. Complete resection is desirable but should not be pursued at the cost of vital organ sacrifice or excessive morbidity. Following surgery, radiation therapy (typically 21.6 Gy in 12 fractions) is delivered to the primary tumor bed and any residual MIBG-avid sites identified on post-induction imaging.
High Risk — Consolidation
High-dose chemotherapy followed by autologous stem cell rescue (ASCT) — using peripheral blood stem cells collected before induction chemotherapy — is the consolidation standard for high-risk disease. The landmark COG ANBL12P1 trial demonstrated that tandem (sequential double) ASCT using carboplatin/etoposide/melphalan for the first transplant and thiotepa/cyclophosphamide for the second significantly improved 3-year EFS compared to single ASCT (61.4% vs 48.4%).
High Risk — Maintenance and Immunotherapy
The most significant advance in high-risk neuroblastoma treatment in the past two decades has been the addition of immunotherapy maintenance after consolidation. The pivotal COG ANBL0032 trial (Yu et al., NEJM 2010) demonstrated that dinutuximab (anti-GD2 chimeric monoclonal antibody, targeting the disialoganglioside GD2 expressed on neuroblastoma cells) combined with GM-CSF, interleukin-2, and isotretinoin (13-cis-retinoic acid) significantly improved 2-year EFS (66% vs 46%) and overall survival compared to isotretinoin alone in patients who had achieved at least a partial response to induction. This immunotherapy combination is now the standard maintenance regimen in North America. Isotretinoin (13-cis-retinoic acid), given for 6 months, promotes differentiation of residual neuroblastoma cells toward a more benign phenotype, reducing the likelihood of relapse from minimal residual disease.
Relapsed and Refractory Disease
Relapsed or refractory high-risk neuroblastoma carries a dismal prognosis, with fewer than 10% of patients achieving long-term survival. Treatment options include:
- ALK inhibitors: Crizotinib (Phase 1-2) and lorlatinib (Phase 3 COG ANBL1232) for ALK-mutated tumors
- I-131 MIBG targeted radiotherapy: Delivers high-dose radiation specifically to MIBG-avid tumor sites; approximately 30% objective response rate as single agent in relapsed disease
- DFMO (difluoromethylornithine): An ornithine decarboxylase inhibitor that blocks polyamine synthesis; being evaluated as maintenance therapy in the Phase 3 PREVENT-NB trial
- Immunotherapy combinations: Anti-GD2 antibody combinations with checkpoint inhibitors, cytokines, and other immunomodulatory agents
- Clinical trials: Enrollment in a clinical trial is the recommended approach for all patients with relapsed or refractory disease
Prognosis
Neuroblastoma outcomes vary enormously based on risk group, reflecting the biological heterogeneity of the disease.
Overall Survival by Risk Group
- All stages combined: Approximately 75% five-year overall survival
- Very low and low risk: Five-year OS exceeds 95%; many patients are cured with surgery alone or minimal treatment
- Intermediate risk: Five-year OS approximately 90 to 95% with risk-adapted chemotherapy
- High risk: Five-year OS has improved from less than 30% in the 1990s to approximately 50 to 60% in the modern immunotherapy era, though long-term cure remains elusive for many patients
High-Risk Disease — Progress and Remaining Challenges
The introduction of tandem ASCT and dinutuximab-based immunotherapy maintenance has been transformative for high-risk neuroblastoma, but relapse remains the primary cause of death. Approximately 50% of high-risk patients who achieve remission after first-line therapy eventually relapse, and the vast majority of those who relapse will die of their disease. Relapsed high-risk neuroblastoma — particularly after two lines of therapy — has fewer than 10% long-term survival, making prevention of relapse the most critical treatment goal.
MYCN Amplification
MYCN amplification confers poor prognosis across all stages. Even infants with otherwise favorable biologic features (low stage, favorable histology) have significantly worse outcomes if their tumor is MYCN-amplified, mandating treatment escalation to high-risk protocols regardless of clinical stage.
Infants with MS Disease — Spontaneous Regression
Infants younger than 18 months with Stage MS disease (skin, liver, and/or bone marrow metastases without cortical bone involvement) who lack unfavorable molecular features have an excellent prognosis, with observed spontaneous regression rates approaching 90% in some series. This regression likely reflects programmed developmental apoptosis of immature neuroblasts when the underlying differentiation signal (presumably embryonic growth factors) is withdrawn postnatally. The ability of a malignant tumor to regress spontaneously without treatment remains one of the most remarkable phenomena in all of oncology.
OMA Syndrome — Neurological Prognosis
Patients with OMA syndrome face a paradox: their tumors tend to be localized and carry better oncological prognoses, but 50 to 70% develop long-term neurodevelopmental sequelae — cognitive impairment, language delay, sleep disorders, behavioral problems, and reduced quality of life — despite successful tumor treatment. The ongoing immune-mediated cerebellar injury, not the tumor itself, drives these outcomes. Immunosuppressive therapy (ACTH, corticosteroids, rituximab) can improve acute OMA symptoms but may not fully prevent long-term neurological damage.
Late Effects of Treatment
Survivors of high-risk neuroblastoma face a significant burden of late treatment effects:
- Hearing loss: Carboplatin and cisplatin cause sensorineural hearing loss; monitoring with serial audiograms is essential
- Scoliosis and musculoskeletal: Radiation to the spine in young children disrupts vertebral growth, causing scoliosis and shortened trunk height
- Growth impairment: High-dose chemotherapy and radiation affect growth hormone axis function
- Cardiac toxicity: Anthracyclines (doxorubicin) cause dose-dependent cardiotoxicity, including cardiomyopathy detectable years after treatment
- Endocrine dysfunction: Hypothyroidism, premature ovarian insufficiency, and adrenal insufficiency occur depending on radiation fields
- Secondary malignancies: Alkylating agents and topoisomerase II inhibitors carry a small but real risk of secondary leukemia; radiation increases risk of secondary solid tumors in the field
- Dinutuximab-related: Severe pain (GD2 is expressed on peripheral nerve fibers), capillary leak syndrome, and neuropathy during infusion are common acute toxicities requiring opioid analgesia
Emerging Therapies and Research
Neuroblastoma research is among the most active areas of pediatric oncology, driven by the large unmet need in high-risk and relapsed disease.
Dinutuximab Beta and Next-Generation Anti-GD2 Antibodies
Dinutuximab beta (ch14.18/CHO), the European variant of the anti-GD2 antibody used by the SIOPEN consortium, differs from North American dinutuximab (ch14.18/SP2/0) in its production cell line and glycosylation pattern. SIOPEN trials have evaluated long-duration subcutaneous infusion regimens that reduce the peak serum concentration and thereby diminish the severe pain associated with intravenous infusion, potentially improving tolerability and outpatient delivery. Next-generation anti-GD2 antibodies and antibody-drug conjugates are in early clinical development.
Lorlatinib (ALK Inhibitor) — Phase 3 Trial
Lorlatinib, a third-generation ALK/ROS1 inhibitor with CNS penetration, is being evaluated in the Phase 3 COG ANBL1232 trial for newly diagnosed high-risk ALK-mutated neuroblastoma. Lorlatinib's CNS penetration addresses a potential sanctuary site and overcomes resistance mutations that limit earlier-generation ALK inhibitors. Results of this trial are anticipated to define the role of lorlatinib in frontline high-risk therapy.
I-131 MIBG Therapy
Therapeutic I-131 MIBG (high-dose targeted radiotherapy using I-131-labeled MIBG) delivers cytotoxic radiation directly to MIBG-avid tumor cells while sparing normal tissues. It achieves approximately 30% objective response rates in relapsed/refractory neuroblastoma and is increasingly being evaluated as a component of frontline consolidation therapy. Iobenguane I-131 (AZEDRA) received FDA approval for adults with pheochromocytoma/paraganglioma, and pediatric neuroblastoma trials are ongoing.
CAR-T Cell Therapy Targeting GD2
Chimeric antigen receptor (CAR) T-cells engineered to target GD2 have entered early-phase clinical trials for neuroblastoma. Early results from trials at Great Ormond Street and other centers show feasibility and early evidence of antitumor activity. Challenges include on-target/off-tumor toxicity (GD2 expression on peripheral nerves causing pain syndromes), T-cell exhaustion in the immunosuppressive tumor microenvironment, and tumor heterogeneity in GD2 expression. Armored CAR-T constructs with IL-15 co-expression to improve persistence are in development.
DFMO Maintenance
Difluoromethylornithine (DFMO, eflornithine), an irreversible inhibitor of ornithine decarboxylase (ODC1), depletes polyamines — molecules essential for cell proliferation that are frequently overproduced in MYCN-amplified neuroblastoma. A single-arm Phase 2 study (NANT 2011-01) suggested improved 2-year EFS with extended DFMO maintenance after first-line therapy. The Phase 3 PREVENT-NB trial (COG ANBL1821) is currently evaluating DFMO as post-immunotherapy maintenance in high-risk patients; results are eagerly awaited.
Peptide Receptor Radionuclide Therapy (PRRT)
Lu-177-DOTATATE and related PRRT agents target somatostatin receptor-expressing tumors. Neuroblastoma cells variably express somatostatin receptors, and PRRT is being evaluated in small studies of MIBG non-avid or MIBG-refractory neuroblastoma, leveraging the complementary targeting mechanism.
International Collaboration
The rarity of neuroblastoma (fewer than 800 cases per year in the USA) makes international collaboration essential for powering randomized trials. The COG Neuroblastoma Committee (North America), SIOPEN (Europe), GPOH (Germany), and JNBSG (Japan) have been harmonizing risk stratification and trial design through the INRG Task Force, enabling larger and more definitive trials than any single country could conduct independently. Genomic profiling platforms such as the COG Pediatric Cancer Data Commons and the SIOPEN BioBank are centralizing molecular data to accelerate biomarker-driven treatment individualization.
Key Research Papers
-
Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010.
Search PubMed — Comprehensive review of neuroblastoma biology, risk stratification, and treatment advances, covering MYCN amplification, genomic markers, and multimodal therapy. -
Cohn SL et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009.
Search PubMed — Landmark paper defining the pretreatment INRG staging system and risk classification framework using image-defined risk factors (IDRFs) and pre-treatment molecular features. -
Brodeur GM et al. Molecular basis for heterogeneity in human neuroblastomas. Eur J Cancer. 1995.
Search PubMed — Foundational analysis of the molecular underpinnings of neuroblastoma heterogeneity including MYCN amplification, ploidy, and chromosomal deletions as biological determinants of clinical behavior. -
Matthay KK et al. Neuroblastoma. Nat Rev Dis Primers. 2016.
Search PubMed — Authoritative disease primer covering epidemiology, pathogenesis, genomic landscape, clinical presentation, staging, treatment paradigms, and future directions. -
Yu AL et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010.
Search PubMed — Pivotal Phase 3 randomized trial demonstrating that dinutuximab-based immunotherapy maintenance significantly improved event-free and overall survival in high-risk neuroblastoma, establishing the current standard of care. -
Park JR et al. Effect of tandem autologous stem cell transplant vs single transplant on event-free survival in patients with high-risk neuroblastoma. JAMA. 2019.
Search PubMed — COG randomized trial showing that tandem ASCT significantly improved 3-year EFS compared to single ASCT in high-risk neuroblastoma, supporting escalated consolidation. -
Pinto NR et al. Advances in risk classification and treatment strategies for neuroblastoma: a report from the Children's Oncology Group and the International Neuroblastoma Risk Group. J Clin Oncol. 2015.
Search PubMed — Comprehensive review of COG and INRG risk classification evolution, contemporary treatment strategies, and survival outcomes across risk groups. -
Attiyeh EF et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005.
Search PubMed — Large retrospective study establishing 1p deletion and 11q deletion as independent prognostic markers in neuroblastoma, with 11q deletion identifying a high-risk subset lacking MYCN amplification. -
Molenaar JJ et al. Sequencing of neuroblastoma identifies chromothripsis and defects that target TERT, a telomerase subunit. Nat Genet. 2012.
Search PubMed — Genomic sequencing study identifying TERT rearrangements and chromothripsis as mechanisms of telomere lengthening in high-risk neuroblastoma, defining the telomere maintenance subgroup. -
Twist CJ et al. Guideline for the treatment of neuroblastoma. Pediatr Blood Cancer. 2019.
Search PubMed — Clinical practice guidelines summarizing evidence-based recommendations for neuroblastoma diagnosis, risk stratification, and treatment across all risk groups. -
Ladenstein R et al. Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma. Lancet Oncol. 2017.
Search PubMed — SIOPEN HR-NBL1 randomized trial comparing two high-dose chemotherapy consolidation regimens in high-risk neuroblastoma, providing European data on optimal ASCT conditioning. -
London WB et al. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children's Oncology Group. J Clin Oncol. 2005.
Search PubMed — Statistical analysis demonstrating that an 18-month (547 days) age cutoff rather than 12 months more accurately predicts outcome in Stage M neuroblastoma, informing the INRG age threshold for risk classification.
Additional PubMed searches: Neuroblastoma treatment MYCN amplification neuroblastoma Dinutuximab neuroblastoma immunotherapy High-risk neuroblastoma stem cell transplant