Spinal Muscular Atrophy (SMA)
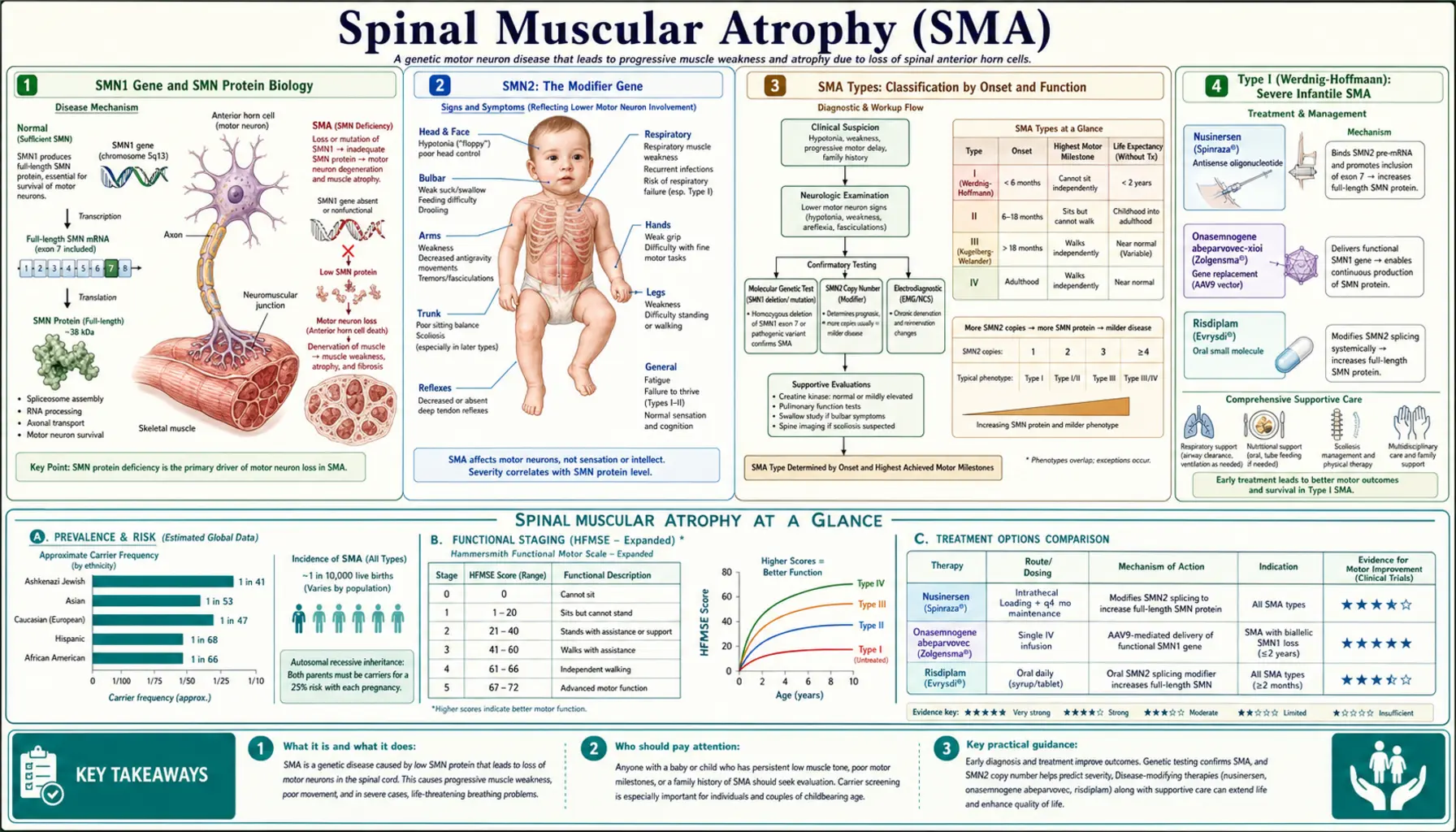
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease caused by deletion or mutation of the SMN1 gene, leading to insufficient survival motor neuron (SMN) protein and progressive degeneration of lower motor neurons in the anterior horn of the spinal cord. The result is relentless proximal muscle weakness that spares cognition entirely. Historically the most common genetic cause of infant death, SMA has been transformed in the past decade by three approved disease-modifying therapies — nusinersen, onasemnogene abeparvovec, and risdiplam — and by universal newborn screening that enables pre-symptomatic treatment before neurons are lost.
- SMN1 Gene and SMN Protein Biology
- SMN2: The Modifier Gene
- SMA Types: Classification by Onset and Function
- Type I (Werdnig-Hoffmann): Severe Infantile SMA
- Types II and III: Intermediate and Mild Forms
- Type IV: Adult-Onset SMA
- Clinical Features: Motor Neuron Degeneration
- Diagnosis: Genetic Testing and EMG
- Newborn Screening
- Nusinersen (Spinraza): Antisense Oligonucleotide Therapy
- Onasemnogene Abeparvovec (Zolgensma): Gene Therapy
- Risdiplam (Evrysdi): Oral SMN2 Splicing Modifier
- Key Research Papers
- Connections
SMN1 Gene and SMN Protein Biology
The SMN1 gene (survival motor neuron 1) sits in the telomeric region of chromosome 5q13 and encodes the 294-amino-acid SMN protein. SMN is a ubiquitously expressed protein that plays a fundamental role in the assembly of spliceosomal small nuclear ribonucleoproteins (snRNPs), the molecular machines that splice pre-mRNA. Motor neurons of the spinal cord anterior horn are exquisitely sensitive to SMN deficiency — a vulnerability whose full mechanistic basis is still under investigation but likely relates to the enormous translational demands placed on these large cells and to SMN's additional roles in axonal mRNA transport.
In the vast majority of SMA cases (approximately 95%), both copies of SMN1 are deleted — either a homozygous deletion of exon 7 and exon 8 or a deletion of exon 7 alone. The remaining ~5% carry a deletion on one allele and a point mutation on the other. Heterozygous carriers (one functional SMN1 copy) are clinically unaffected; carrier frequency in the general population is approximately 1 in 40–50, making SMA incidence roughly 1 in 6,000–10,000 live births.
When SMN protein falls below a critical threshold during spinal cord development and postnatal maturation, anterior horn motor neurons die through a process that is not fully reversible. The degenerative process begins before birth in severe forms, which is why pre-symptomatic intervention — enabled by newborn screening — yields dramatically better outcomes than treatment initiated after weakness appears. Once a motor neuron pool is depleted, even the most effective SMN-restoring therapies cannot regenerate it.
Beyond its canonical snRNP assembly role, SMN protein participates in mRNA splicing regulation, stress granule dynamics, and the trafficking of beta-actin mRNA to growth cones. Deficiency therefore disrupts multiple pathways simultaneously, and understanding which defects matter most in motor neurons remains an active research frontier that could reveal additional therapeutic targets.
Back to Table of ContentsSMN2: The Modifier Gene
Immediately centromeric to SMN1 on chromosome 5q13 lies SMN2, an inverted duplication of SMN1 that differs by only a handful of nucleotides. The critical difference is a C-to-T transition at position 6 of exon 7. This single nucleotide change disrupts an exonic splicing enhancer and creates an exonic splicing silencer, causing the spliceosome to skip exon 7 approximately 85–90% of the time during SMN2 pre-mRNA processing. The resulting truncated transcript (lacking exon 7) encodes a protein, SMNΔ7, that is unstable and rapidly degraded. Only about 10–15% of SMN2 transcripts include exon 7 and generate full-length, functional SMN protein.
This inefficient production of full-length SMN from SMN2 is the reason SMA patients survive at all — complete absence of both SMN1 and SMN2 is embryonic lethal in mice. The number of SMN2 gene copies a patient carries is the primary modifier of disease severity: individuals with two copies produce less full-length SMN and typically develop type I SMA; those with three copies tend toward type II; those with four or more copies often present with type III or even type IV. This genotype-phenotype correlation is powerful but not absolute — other genetic and epigenetic modifiers exist, including plastin-3 overexpression, which protects some individuals with few SMN2 copies from the most severe forms.
The SMN2 gene became the central therapeutic target once researchers recognized that correcting the exon 7 splicing defect would restore adequate full-length SMN production. All three approved therapies exploit this biology: nusinersen and risdiplam promote SMN2 exon 7 inclusion by different molecular mechanisms, while onasemnogene abeparvovec delivers a functional SMN1 transgene to bypass the problem entirely.
Back to Table of ContentsSMA Types: Classification by Onset and Function
SMA is classified into types 0 through IV based on age of onset and the highest motor milestone achieved. This classification predates genetic testing and reflects the clinical spectrum that emerges from varying degrees of SMN protein deficiency. Types I through IV account for almost all clinically recognized cases, with type I (also called Werdnig-Hoffmann disease) being the most common, comprising roughly 60% of cases. The classification remains clinically useful for prognosis and treatment planning, though it is increasingly supplemented by genetic data (SMN2 copy number) and by the emerging category of "pre-symptomatic SMA" identified through newborn screening before any milestone failure occurs.
Type 0, the most severe form, presents prenatally with reduced fetal movements, arthrogryposis (joint contractures from lack of movement in utero), and respiratory failure at birth. Infants with type 0 rarely survive more than weeks and almost always carry only one SMN2 copy. Type I is the classic infantile form with onset before 6 months; type II presents in the first 18 months with intermediate severity; type III (Kugelberg-Welander) presents after 18 months with mild-to-moderate weakness; type IV presents in adulthood with minimal disability.
It is important to understand that these types describe untreated natural history. Children identified pre-symptomatically through newborn screening and treated before significant motor neuron loss can achieve motor milestones far beyond what their genotype would historically have predicted. The transformation of SMA's natural history by early treatment has blurred the traditional type boundaries and raised the possibility that SMA in the newborn screening era will become a treatable chronic condition rather than an inevitably fatal or profoundly disabling one.
Back to Table of ContentsType I (Werdnig-Hoffmann): Severe Infantile SMA
Type I SMA presents within the first 6 months of life with the hallmark picture of a profoundly hypotonic infant who cannot achieve or maintain head control and will never sit independently without treatment. Parents often describe their baby as unusually floppy from birth, and newborns may show reduced spontaneous movements, a weak cry, and poor feeding. On examination the infant is limp ("rag-doll" hypotonia), with absent or markedly diminished deep tendon reflexes, visible tongue fasciculations (a nearly pathognomonic finding in an infant), and a distinctive bell-shaped chest resulting from intercostal muscle weakness with relative sparing of the diaphragm.
The respiratory picture is critical and life-limiting in untreated type I SMA. Because the intercostal muscles weaken faster than the diaphragm, affected infants breathe paradoxically — the chest wall collapses inward during inspiration instead of expanding. This inefficient breathing pattern leads to hypoventilation, recurrent aspiration pneumonia, and respiratory failure. In the pre-treatment era, more than 90% of type I infants died or required permanent ventilation before the age of 2 years. The bulbar muscles are also affected, causing swallowing dysfunction that compounds respiratory risk and impairs adequate nutrition.
The cognitive, sensory, and autonomic systems are completely spared in SMA — these children are alert, engaged, emotionally responsive, and intellectually normal. The devastating contrast between an intact mind and a body unable to move or breathe is one of the most poignant features of the disease and has driven the extraordinary urgency with which families, clinicians, and researchers pursued treatment development. Despite intact cognition, the combination of immobility, respiratory failure, and feeding difficulty historically made survival beyond early childhood exceptional without aggressive ventilatory and nutritional support.
Most type I patients carry two copies of SMN2, though a subset with three copies can have a slightly milder course (sometimes called type I.5 or "intermediate type I"). Genetic confirmation of the SMN2 copy number at diagnosis helps guide treatment urgency and prognosis, particularly in determining eligibility for onasemnogene abeparvovec, which is FDA-approved for patients under 2 years of age.
Back to Table of ContentsTypes II and III: Intermediate and Mild Forms
Type II SMA (also called chronic infantile or Dubowitz disease) presents between 7 and 18 months of age. Affected children achieve the ability to sit independently — the defining milestone — but never walk unaided. Motor development is initially normal or near-normal and then plateaus or slowly regresses as the anterior horn cell population continues to decline. Fine motor skills are preserved longer than gross motor skills, and these children often remain cognitively, socially, and emotionally engaged well into adulthood. The typical SMN2 copy number is three.
The major long-term complications of type II SMA are scoliosis and respiratory compromise. Loss of truncal muscle support leads to progressive scoliotic curves that can reach severe angles, compressing the chest wall and further impairing breathing. Spinal fusion surgery is often required but carries respiratory risk. Nocturnal hypoventilation typically precedes daytime respiratory failure and is managed with non-invasive ventilation (BiPAP). With careful respiratory monitoring and support, type II patients can survive into adulthood, though life expectancy remains reduced compared to the general population in the absence of disease-modifying therapy.
Type III SMA (Kugelberg-Welander disease) presents after 18 months — by definition, affected children have walked independently at some point. Type IIIa presents before age 3; type IIIb presents between ages 3 and 18. Early in the disease, weakness is proximal and lower-extremity predominant — a Gowers' sign (using hands to push up from the floor) is often noted. Ambulation is eventually lost in many patients, particularly those with type IIIa, though the timeline varies from childhood to late adulthood. Type IIIb patients often retain the ability to walk well into their fifties or beyond. Life expectancy in type III is generally normal or near-normal, and respiratory involvement is mild in most cases. Most type III patients carry three to four copies of SMN2.
Back to Table of ContentsType IV: Adult-Onset SMA
Type IV SMA manifests in adulthood, typically after age 30, and follows a slowly progressive course that has minimal impact on life expectancy. Patients present with proximal lower extremity weakness, difficulty climbing stairs, and occasional falls. The pattern is symmetric and proximal, mimicking limb-girdle muscular dystrophy or other late-onset neuromuscular diseases, and the diagnosis is often delayed for years until genetic testing is performed. Type IV patients typically carry four or more copies of SMN2, which provides enough full-length SMN to sustain motor neurons through decades of postnatal life.
Because type IV presents so subtly and so late, it was historically underdiagnosed. The widespread availability of comprehensive neuromuscular gene panels has improved diagnostic accuracy, and many adults with a diagnosis of "atypical limb-girdle dystrophy" or "adult-onset proximal myopathy" are now correctly identified as having SMA type IV. Awareness of the diagnosis matters because disease-modifying therapies are approved across SMA types, not just in childhood-onset disease, and because accurate genetic counseling depends on correct diagnosis — siblings and children of type IV patients may carry heterozygous SMN1 deletions that carry reproductive implications.
Back to Table of ContentsClinical Features: Motor Neuron Degeneration
The core clinical signature of SMA is symmetric proximal muscle weakness that is greater in the legs than in the arms and involves lower motor neurons without upper motor neuron signs. Deep tendon reflexes are absent or markedly diminished (areflexia) — a cardinal feature that distinguishes SMA from myopathies, which preserve reflexes until very late. Muscle bulk is reduced from denervation atrophy, and fasciculations (spontaneous motor unit discharges visible under the skin) are present, particularly in the tongue, which is easily examined in infants. The proximal-greater-than-distal pattern and the leg-greater-than-arm pattern reflect the selectivity with which anterior horn cells degenerate in SMA — the reason is not fully understood but may relate to the size and metabolic demands of different motor neuron pools.
A critical clinical teaching point is what SMA does not affect. Cognition, language, memory, and personality are entirely normal at all ages — affected children often show remarkable intellectual precocity, a feature that clinicians should explicitly discuss with families to avoid the mistaken assumption that motor severity implies cognitive impairment. Sensation is also completely spared, distinguishing SMA from Charcot-Marie-Tooth disease and other hereditary motor and sensory neuropathies. There are no upper motor neuron signs (spasticity, Babinski sign, hyperreflexia), distinguishing SMA from ALS in adults. Sphincter function and autonomic control are normal. Eye movements are normal. Cardiac involvement is not a primary feature, though some patients with severe forms develop heart defects that may represent a separate manifestation of SMN deficiency in cardiac tissue.
Breathing mechanics in SMA deserve special attention. The intercostal muscles (which expand the ribcage) weaken before the diaphragm (which pulls the chest down), creating the characteristic bell-shaped chest and paradoxical breathing pattern described under type I. This intercostal-before-diaphragm hierarchy means that standard pulmonary function tests can underestimate respiratory compromise — maximum inspiratory and expiratory pressures and nocturnal oximetry are more informative than simple spirometry in non-ambulatory patients. Cough effectiveness depends on expiratory muscle strength; when cough is weak, secretions accumulate and pneumonia risk rises dramatically. Assisted cough techniques and airway clearance devices are standard components of SMA management.
Scoliosis and joint contractures develop in non-ambulatory patients as a consequence of imbalanced muscle forces and chronic positioning. In type II patients, spinal curves can progress rapidly during the pubertal growth spurt, and surgical management requires careful pre-operative respiratory assessment because many of these patients cannot tolerate the post-operative respiratory demands of thoracic surgery without non-invasive ventilatory support. Hip dislocation from hypotonia is common in non-ambulatory infants. Nutritional management is also a major clinical challenge: bulbar weakness impairs swallowing safety and caloric intake, frequently requiring nasogastric or gastrostomy tube feeding in type I and severe type II patients.
Back to Table of ContentsDiagnosis: Genetic Testing and EMG
The definitive diagnosis of SMA rests on molecular genetic testing demonstrating deletion or mutation of both copies of the SMN1 gene. Multiplex ligation-dependent probe amplification (MLPA) is the standard first-line test; it reliably detects homozygous deletion of SMN1 exon 7, which accounts for ~95% of cases, and simultaneously quantifies SMN2 copy number. When MLPA shows only one copy of SMN1 (suggesting compound heterozygosity rather than homozygous deletion), sequencing of the intact allele is performed to identify the second pathogenic variant. The turn-around time for MLPA-based SMA diagnosis is typically 2–5 days in clinical laboratories, which is critical given the urgency of initiating treatment in type I infants.
Electromyography (EMG) and nerve conduction studies (NCS) can support the diagnosis before genetic testing is complete and remain useful when the genetic picture is ambiguous. EMG shows a neurogenic pattern — increased insertional activity, fibrillation potentials, positive sharp waves (signs of active denervation), large-amplitude long-duration polyphasic motor unit action potentials (signs of chronic reinnervation), and reduced recruitment. Nerve conduction studies show normal sensory nerve action potentials and normal or near-normal motor nerve conduction velocities with reduced compound muscle action potential amplitudes — a pattern consistent with anterior horn cell disease rather than peripheral nerve disease. This NCS pattern, where sensory nerves are spared and motor velocity is preserved (unlike the slowed velocities in demyelinating CMT), is an important distinguishing feature.
Serum creatine kinase (CK) is typically normal or mildly elevated in SMA, in contrast to the markedly elevated CK seen in muscular dystrophies. Muscle biopsy is rarely needed for diagnosis in the current era of genetic testing but would show a characteristic pattern of grouped atrophy (large fascicles of atrophic fibers from denervation adjacent to hypertrophied fibers from compensatory reinnervation) rather than the random fiber atrophy seen in myopathies. Prenatal diagnosis is available through chorionic villus sampling or amniocentesis for families with a known SMA diagnosis. Carrier testing of at-risk family members using MLPA is accurate for detecting the common deletion but has limitations: it cannot distinguish individuals with one SMN1 copy from those with two copies on the same chromosome (2+0 carrier configuration), and carrier testing should be accompanied by genetic counseling that explains these limitations.
Back to Table of ContentsNewborn Screening
SMA was added to the Recommended Uniform Screening Panel (RUSP) for US newborn screening in 2018, following evidence that pre-symptomatic treatment dramatically alters disease course. The standard newborn screen for SMA uses a dried blood spot (heel prick sample, collected at 24–48 hours of life) and real-time quantitative PCR to detect homozygous deletion of SMN1 exon 7. The test is highly sensitive and specific for the ~95% of SMA cases caused by homozygous deletion; compound heterozygotes with one deletion and one point mutation are not reliably detected by the screen and may still present clinically.
The impact of newborn screening on outcomes has been remarkable. In the pre-symptomatic cohort of the NURTURE trial (nusinersen initiated before 6 weeks of age in infants carrying 2–3 SMN2 copies), 91% of children sat independently and 73% walked independently at 3 years — milestone achievements that were virtually unknown in untreated type I and II natural history. Similarly, infants treated with onasemnogene abeparvovec pre-symptomatically in the SPR1NT trial achieved age-appropriate motor milestones indistinguishable from unaffected peers at 18 months. These data firmly established that the window for maximal therapeutic benefit is before, not after, motor neuron depletion begins.
Not all US states implemented SMA newborn screening simultaneously after the 2018 RUSP addition — implementation required state legislation and laboratory infrastructure. By 2022 the large majority of US states had programs in place. Internationally, SMA newborn screening programs are expanding rapidly in Europe, Asia, and Latin America, driven by the availability of approved therapies and the compelling evidence that early treatment changes lives. When a positive screen result is returned, the standard workflow is urgent SMN2 copy number quantification followed by immediate referral to a neuromuscular center for treatment initiation — with the goal of beginning therapy before any motor symptoms appear, ideally within the first weeks of life.
Back to Table of ContentsNusinersen (Spinraza): Antisense Oligonucleotide Therapy
Nusinersen (brand name Spinraza, developed by Ionis Pharmaceuticals and marketed by Biogen) was approved by the FDA in December 2016 — the first disease-modifying therapy for SMA and a landmark moment for the entire antisense oligonucleotide (ASO) field. Nusinersen is a chemically modified 18-nucleotide ASO that binds to intronic splicing silencer N1 (ISS-N1) in the SMN2 pre-mRNA, blocking the splicing repressor that normally causes exon 7 to be skipped. By occupying ISS-N1, nusinersen shifts SMN2 splicing toward exon 7 inclusion, increasing the proportion of full-length, functional SMN protein produced from SMN2 copies. This approach elegantly harnesses the endogenous gene the patient already carries rather than delivering exogenous genetic material.
Because nusinersen does not cross the blood-brain barrier after systemic administration, it is delivered by intrathecal injection — a lumbar puncture that deposits the drug directly into the cerebrospinal fluid, from which it distributes throughout the spinal cord and brain. The dosing regimen involves four loading doses (at days 0, 14, 28, and 63) followed by maintenance doses every 4 months for the life of the patient. Each intrathecal injection requires sedation or careful positioning in infants, and patients with significant scoliosis may require fluoroscopic or ultrasound guidance. The procedure is generally well tolerated; the main adverse effects are those associated with lumbar puncture itself (post-procedural headache, transient back pain, and a small risk of infection).
The pivotal ENDEAR trial enrolled type I infants and demonstrated that nusinersen significantly improved motor milestone achievement and event-free survival (freedom from permanent ventilation or death) compared to sham procedure. Approximately 41% of nusinersen-treated infants achieved motor milestones (vs. 0% of controls), and event-free survival was markedly better in the treated group. The CHERISH trial in type II/III patients demonstrated improvements in motor function scores on the Hammersmith Functional Motor Scale. Long-term open-label extension data continue to show sustained benefit across SMA types, and real-world registries confirm that nusinersen is effective even when initiated in older symptomatic patients, though outcomes are best when treatment begins early.
Nusinersen is approved across all SMA types and ages with no upper age limit, making it the only one of the three approved therapies available to older children and adults who are not candidates for gene therapy. Its intrathecal route is a practical challenge, particularly in patients with severe scoliosis who have undergone spinal fusion — accessing the intrathecal space through fused hardware requires specialist interventional radiology in some cases — but the route also ensures concentrated CNS delivery without systemic exposure. The cost of nusinersen is approximately $125,000 per maintenance injection, making it among the most expensive drugs per year in the world, and access disparities remain a significant global public health concern.
Back to Table of ContentsOnasemnogene Abeparvovec (Zolgensma): Gene Therapy
Onasemnogene abeparvovec (brand name Zolgensma, developed by AveXis and marketed by Novartis Gene Therapies) was approved by the FDA in May 2019 for patients under 2 years of age with SMA — the first approved gene therapy for a neuromuscular disease and one of the most expensive single-dose treatments in medical history at approximately $2.1 million USD. The therapy uses a self-complementary adeno-associated virus serotype 9 (scAAV9) vector to deliver a functional copy of the human SMN1 gene under the control of the chicken beta-actin hybrid (CBh) promoter. AAV9 is chosen for its ability to efficiently transduce motor neurons in the spinal cord after intravenous administration, owing to the natural tropism of AAV9 for the central nervous system.
The drug is administered as a single intravenous infusion over approximately one hour. AAV9 vectors do not integrate into the host genome in a targeted manner — they persist predominantly as non-replicating episomal DNA in the nucleus of transduced cells. Because motor neurons are post-mitotic (they do not divide), the episomal transgene is not diluted by cell division and can provide long-term expression. However, because the episome is lost if a cell divides, the therapy is most effective when delivered early in infancy before somatic growth substantially dilutes the proportion of transfected cells. This biological rationale underpins the age restriction.
The pivotal STR1VE trial demonstrated that 91% of treated type I infants achieved and maintained the ability to sit without support for at least 30 seconds by 18 months of age, compared to the historical natural history in which virtually no type I infants achieved this milestone. In the pre-symptomatic SPR1NT trial, all infants with two SMN2 copies achieved independent sitting, 9 of 10 achieved independent standing, and all enrolled participants with two or three SMN2 copies achieved independent walking at 14 months. Two-year follow-up data continue to show durability of benefit, though the question of whether a single dose will provide lifelong protection or require re-dosing eventually remains under investigation.
The major safety considerations for onasemnogene abeparvovec are immune-mediated hepatotoxicity (a transaminase elevation that peaks around 2–3 weeks post-infusion and requires pre-emptive oral prednisolone prophylaxis) and thrombotic microangiopathy (a rare but serious complication observed in some patients, requiring close platelet and renal monitoring in the first 2 months). Pre-existing antibodies to AAV9 (measured as anti-AAV9 IgG titers) are an exclusion criterion because they prevent vector transduction — patients with high baseline titers cannot receive the therapy. Because natural AAV9 infection is common in childhood, the window for eligibility narrows with age, adding urgency to early administration.
Back to Table of ContentsRisdiplam (Evrysdi): Oral SMN2 Splicing Modifier
Risdiplam (brand name Evrysdi, developed by Roche, Genentech, and the SMA Foundation) received FDA approval in August 2020, providing the first oral disease-modifying therapy for SMA. Risdiplam is a small-molecule compound that acts as an SMN2 pre-mRNA splicing modifier — it binds to and stabilizes the stem-loop structure in SMN2 pre-mRNA at the 5' splice site of exon 7, and also modulates the RNA-binding protein RBM17 to promote exon 7 inclusion. Like nusinersen, risdiplam works by increasing the proportion of full-length SMN protein produced from SMN2 copies, but it achieves this through an entirely different mechanism and, crucially, through systemic oral bioavailability.
Risdiplam is administered as a liquid formulation once daily by mouth (or via feeding tube), making it substantially more convenient than intrathecal nusinersen, particularly for patients with scoliosis who face challenging lumbar puncture access. Because it distributes systemically after oral absorption, risdiplam reaches not only spinal cord motor neurons but also motor neurons in the brainstem, peripheral tissues, and — importantly for SMA biology — potentially the peripheral nervous system and muscle. Whether systemic distribution confers clinical advantages over intrathecal delivery remains an active research question. Risdiplam does cross the placenta and is teratogenic in animal models, so pregnancy is a contraindication.
The pivotal FIREFISH trial in type I infants showed that 29% of treated patients could sit without support for at least 5 seconds at 12 months — a modest-seeming result compared to pre-symptomatic trials but remarkable given that no untreated type I infant historically achieved this milestone. The SUNFISH trial in types II/III patients ages 2–25 demonstrated significant improvement on the Motor Function Measure 32 scale compared to placebo. The RAINBOWFISH trial enrolled pre-symptomatic infants with 1–3 SMN2 copies, and preliminary results mirror the transformative outcomes seen with the other therapies in this earliest intervention window. Risdiplam is approved for patients 2 months of age and older with all SMA types, with no upper age limit, making it a practical option for adult-onset type IV patients as well.
A key practical question in current SMA care is whether patients already receiving nusinersen or who received onasemnogene abeparvovec should switch to or add risdiplam, and whether combination therapy with two SMN-augmenting strategies confers additional benefit. Clinical trials exploring nusinersen plus risdiplam combinations are ongoing. For newly diagnosed patients, the choice among the three therapies depends on age, SMN2 copy number, disease severity, patient and family preference, access/insurance coverage, and institutional experience — decisions best made at specialized SMA centers with multidisciplinary teams.
Back to Table of ContentsKey Research Papers
- Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–165 — Search PubMed
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96(11):6307–6311 — Search PubMed
- Munsat TL, Davies KE. International SMA Consortium Meeting. Neuromuscul Disord. 1992;2(5-6):423–428 — Search PubMed
- Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy (ENDEAR). N Engl J Med. 2017;377(18):1723–1732 — Search PubMed
- Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy (CHERISH). N Engl J Med. 2018;378(7):625–635. PMID: 29091570
- Mendell JR, Al-Zaidy S, Shell R, et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. 2017;377(18):1713–1722. PMID: 30669677
- Glascock J, Sampson J, Connelly S, et al. Revised Recommendations for the Treatment of Infants Diagnosed with Spinal Muscular Atrophy Via Newborn Screening Who Are Pre-symptomatic at Time of Treatment Initiation. J Neuromuscul Dis. 2020;7(2):97–100 — Search PubMed
- Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10(8):597–609 — Search PubMed
- De Vivo DC, Bertini E, Swoboda KJ, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy (NURTURE). Neurology. 2019;93(14):e1417–e1425 — Search PubMed
- Mercuri E, Oskoui M, Muntoni F, et al. Risdiplam in Type 1 Spinal Muscular Atrophy (FIREFISH). N Engl J Med. 2021;384(10):915–923 — Search PubMed
- Strauss KA, Farrar MA, Muntoni F, et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the Phase III SPR1NT trial. Nat Med. 2022;28(7):1381–1389 — Search PubMed
- Kolb SJ, Coffey CS, Yankey JW, et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. 2017;82(6):883–891 — Search PubMed
Connections
- Neurology
- Amyotrophic Lateral Sclerosis (ALS)
- Muscular Dystrophy
- Myotonic Dystrophy (DM1/DM2)
- Myasthenia Gravis
- Lambert-Eaton Myasthenic Syndrome
- Charcot-Marie-Tooth Disease
- Guillain-Barré Syndrome
- Multiple Sclerosis
- Diseases — Main Index