Myotonic Dystrophy (DM1 and DM2)
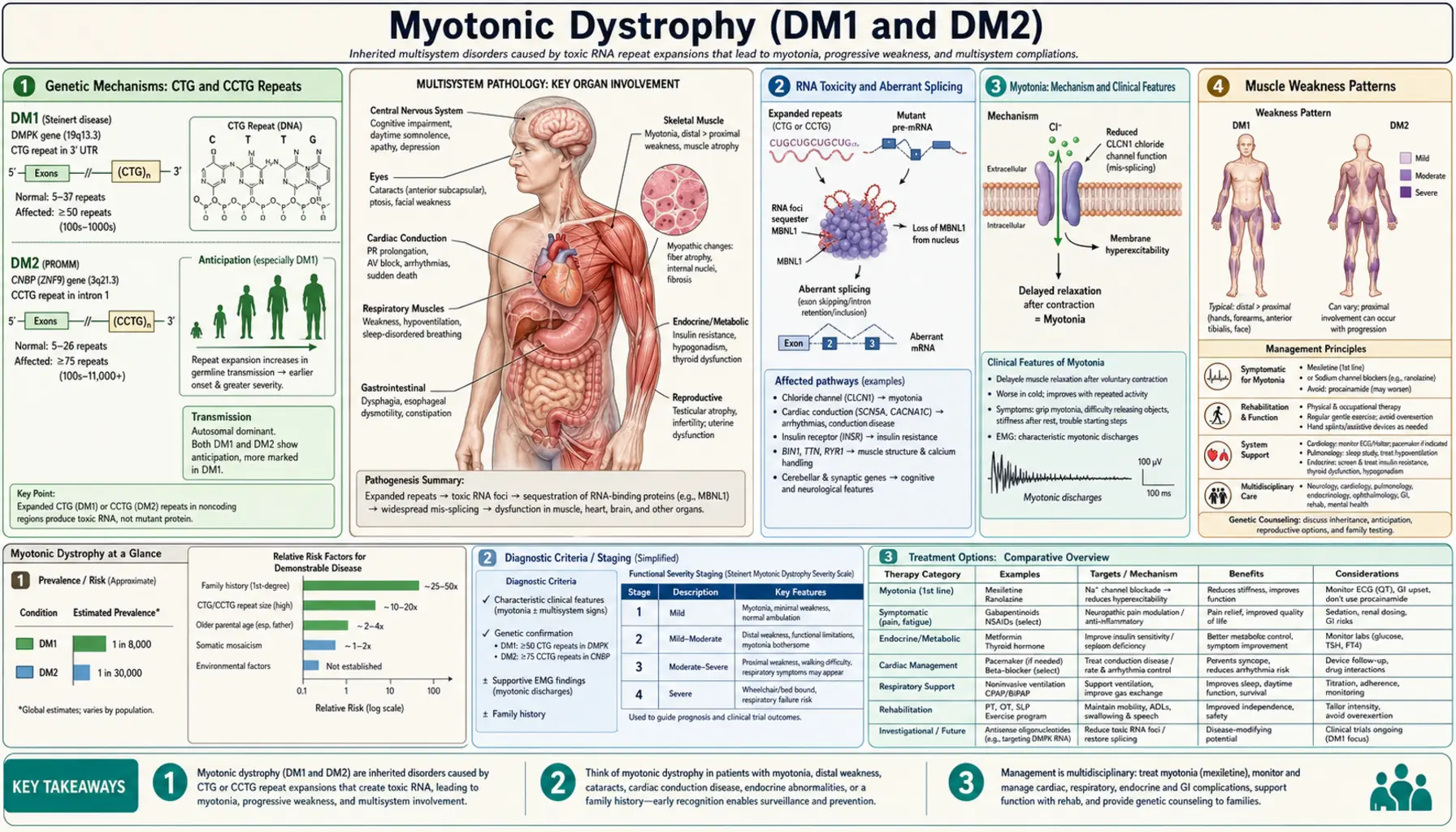
Myotonic dystrophy is the most common adult-onset muscular dystrophy, affecting approximately 1 in 8,000 people worldwide. It is a multisystem disorder caused by toxic RNA produced from expanded nucleotide repeats — not a structural protein defect, but a gene-regulation catastrophe that disrupts the splicing of dozens of transcripts simultaneously. Two genetically distinct forms exist: DM1 (Steinert's disease, CTG expansion in DMPK on chromosome 19) and DM2 (CCTG expansion in CNBP on chromosome 3). Both cause myotonia and progressive muscle weakness, but differ in distribution, severity, and multisystem involvement. Understanding myotonic dystrophy requires grasping why a repeat expansion in a non-coding region can simultaneously affect muscle, heart, brain, eye, and endocrine tissue.
- Genetic Mechanisms: CTG and CCTG Repeats
- RNA Toxicity and Aberrant Splicing
- Myotonia: Mechanism and Clinical Features
- Muscle Weakness Patterns
- Cardiac Complications
- Respiratory and Sleep Involvement
- Brain, Cognition, and Fatigue
- Multisystem: Eye, Endocrine, GI
- DM1 vs DM2 Comparison
- Diagnosis
- Treatment and Management
- Key Research Papers
- Connections
Genetic Mechanisms: CTG and CCTG Repeats
Myotonic dystrophy type 1 (DM1) is caused by an unstable CTG trinucleotide repeat expansion in the 3' untranslated region (UTR) of the DMPK gene (dystrophia myotonica protein kinase) on chromosome 19q13. In the normal population, this CTG repeat is polymorphic but stable, ranging from 5 to 37 copies. A "premutation" range of 38 to 49 repeats may be clinically silent but is prone to expansion in subsequent generations. Classic adult-onset DM1 typically involves 100 to 1,000 CTG repeats, mild DM1 ranges from 50 to 150, and the severe congenital form is almost always associated with more than 1,000 repeats — occasionally reaching tens of thousands.
A defining feature of DM1 is genetic anticipation: the repeat tends to expand with each successive generation, making the disease progressively earlier in onset and more severe in offspring. Crucially, the largest expansions — and therefore congenital DM1 — occur almost exclusively through maternal transmission. A mother carrying a large DM1 expansion is at high risk of transmitting an even larger expansion to her child, sometimes in the congenital range. This is the opposite of the paternal transmission bias seen in Huntington disease, and it has profound implications for genetic counseling of DM1 families.
Myotonic dystrophy type 2 (DM2) is caused by a CCTG tetranucleotide repeat expansion in intron 1 of the CNBP gene (CCHC-type zinc finger, nucleic acid binding protein, formerly known as ZNF9) on chromosome 3q21. Normal individuals carry fewer than 26 CCTG repeats. DM2 patients carry more than 75 repeats and often far more — DM2 repeat expansions commonly reach thousands or even tens of thousands of units. Despite the much larger raw repeat count, DM2 behaves as a milder disease than classic DM1 for reasons not fully understood. Crucially, DM2 does not produce a congenital form and does not show the dramatic anticipation characteristic of DM1, likely because the CCTG expansion does not show the same severe instability during maternal meiosis.
Both mutations are autosomal dominant — a single expanded allele on one chromosome is sufficient to cause disease. Penetrance is high, though variable expressivity means that even within a family, one person may be mildly symptomatic while a sibling with a similar repeat length is more severely affected. The absence of a protein-level defect as the primary mechanism (DMPK haploinsufficiency contributes modestly but is not the main driver) was one of the major surprises of myotonic dystrophy genetics and helped open the entire field of RNA-mediated pathogenesis.
Back to Table of ContentsRNA Toxicity and Aberrant Splicing
The central pathogenic mechanism in myotonic dystrophy is not a defective protein but a toxic RNA. When the DMPK gene is transcribed, the expanded CUG repeats in the mRNA fold into thermodynamically stable hairpin structures. These RNA hairpins accumulate in the nucleus, forming distinct foci visible by fluorescence in situ hybridization (FISH) in patient muscle and brain cells. The hairpins act as molecular sponges, sequestering RNA-binding proteins that are critical for normal gene regulation.
The most important sequestered proteins are MBNL1 (muscleblind-like protein 1) and MBNL2 (muscleblind-like protein 2). MBNL proteins are evolutionarily conserved RNA-binding proteins that normally regulate developmentally programmed alternative splicing — they guide the switch from fetal splice isoforms to adult splice isoforms during postnatal maturation. When MBNL1 and MBNL2 are trapped by the CUG hairpins, this developmental switch fails to complete. Adult tissues in DM1 patients retain fetal splicing patterns for dozens of transcripts, producing what is called a "spliceopathy."
A second pathway amplifies the damage: the expanded CUG RNA aberrantly activates CUGBP1 (also called CELF1, CUG-binding protein 1/CUGBP Elav-like family member 1), partly through PKC-mediated hyperphosphorylation that stabilizes the protein. CUGBP1 is an antagonist of MBNL function at many splice sites, so its upregulation compounds the mis-splicing driven by MBNL sequestration. The net result is that dozens of mis-splicing events occur in parallel across multiple organ systems.
The key downstream targets of mis-splicing explain the clinical syndrome. In skeletal muscle, the adult isoform of the ClC-1 chloride channel (CLCN1) is not produced — instead, a fetal isoform with reduced chloride conductance accumulates, causing membrane hyperexcitability and myotonia. In heart muscle, aberrant splicing of cardiac troponin T (TNNT2) and SCN5A (cardiac sodium channel) contributes to arrhythmias. In brain, mis-splicing of APP, tau, NMDA receptor subunits, and other transcripts contributes to cognitive dysfunction. In the insulin pathway, mis-splicing of the insulin receptor (IR-A vs IR-B isoform shift) contributes to insulin resistance. This elegant mechanistic unification — one mutation, dozens of mis-splicing events, multisystem disease — makes myotonic dystrophy one of the most instructive examples in all of molecular medicine.
Back to Table of ContentsMyotonia: Mechanism and Clinical Features
Myotonia is the hallmark symptom that gives myotonic dystrophy its name. It refers to delayed muscle relaxation after voluntary contraction or mechanical stimulation — the muscle fires repetitively and cannot efficiently return to the relaxed state. Patients experience myotonia as stiffness or difficulty "letting go," most noticeable after sustained or forceful muscle use. The grip myotonia is classic: after firmly shaking hands, the patient cannot quickly release the handshake, and may appear to hold on awkwardly for several seconds. If asked to make a fist and then rapidly open the fingers, the fingers open slowly and incompletely on the first attempt.
Percussion myotonia provides a direct clinical test: tapping the thenar eminence of the palm with a reflex hammer causes the thumb to abduct and adduct back slowly over 5–10 seconds. Percussion over the tongue or deltoid can also elicit a sustained dimple or contraction. This finding is pathognomonic for myotonic disorders and is absent in most other neuromuscular diseases. Importantly, myotonia worsens in cold temperatures, as the membrane changes underlying the condition are temperature-sensitive — patients often notice that their hands stiffen dramatically when reaching into a freezer or working outdoors in winter.
The molecular mechanism of myotonia in DM1 is now well understood. MBNL1 sequestration leads to mis-splicing of CLCN1, the gene encoding the skeletal muscle chloride channel ClC-1. Adult skeletal muscle has abundant ClC-1 activity that normally stabilizes the membrane resting potential after an action potential — chloride conductance constitutes roughly 80% of the resting membrane conductance in mature muscle. When the adult ClC-1 splice isoform is replaced by the low-conductance fetal isoform, resting membrane conductance falls dramatically. After a single action potential, the membrane cannot repolarize efficiently, and repetitive spontaneous action potentials propagate — the electrophysiological basis of myotonia.
On needle EMG, myotonic discharges produce one of the most recognizable sounds in clinical neurophysiology: a waxing-and-waning pattern of electrical discharges that rises and falls in both frequency and amplitude, likened to the sound of a dive-bomber aircraft engine. This characteristic "dive-bomber" sound is heard on EMG even in muscles with no clinically apparent myotonia, making EMG a highly sensitive screen. One important clinical feature of DM1 myotonia is the warm-up phenomenon: myotonia paradoxically improves with repeated exercise. After five or ten attempts, the patient can open the fist much more quickly. This warm-up effect distinguishes DM1 from non-dystrophic myotonias such as myotonia congenita, where repeated activity may not help as consistently.
Back to Table of ContentsMuscle Weakness Patterns
Beyond myotonia, myotonic dystrophy causes progressive muscle weakness and wasting, but the distribution is strikingly different from most muscular dystrophies. In DM1, weakness is distal greater than proximal — the opposite pattern from limb-girdle or Duchenne muscular dystrophy. This distal predominance is highly diagnostically useful and often the first clue that a patient with suspected muscular dystrophy actually has DM1.
The ankle dorsiflexors and toe extensors are affected early and prominently, producing foot drop. Patients develop a characteristic steppage gait — lifting the knee high with each step to prevent the toe from dragging along the floor. Falls are common, especially on uneven surfaces or stairs. Wrist extensors and finger extensors weaken, causing difficulty with fine motor tasks and a tendency to drop objects. As disease progresses, intrinsic hand muscles become involved, further impairing dexterity.
Facial muscle involvement is a defining feature of DM1 that is absent or much milder in most other muscular dystrophies. Bilateral ptosis (drooping eyelids) appears early, sometimes before patients recognize any limb weakness. Temporal muscle wasting creates a characteristic "hatchet face" appearance — the temples are hollowed, the face appears long and narrow, and the jaw may hang slightly open due to masseter weakness. Bilateral facial weakness prevents patients from whistling, puffing out their cheeks, or fully closing their eyes (lagophthalmos). The combination of ptosis and bilateral facial weakness with temporal wasting is nearly pathognomonic for DM1.
Neck flexor weakness — particularly sternocleidomastoid muscle atrophy — is another early finding in DM1. Patients may have difficulty lifting their head off a pillow and develop a slightly forward-flexed neck posture over time. Dysphagia arises from weakness and myotonia affecting pharyngeal and esophageal muscles, and aspiration risk increases as disease progresses. Patients often describe difficulty initiating swallowing or a sense that food "sticks" in the throat or upper chest.
In DM2, the weakness pattern is proximal greater than distal, more closely resembling limb-girdle muscular dystrophy. Hip flexors and thigh muscles (quadriceps) are most affected, causing difficulty rising from chairs, climbing stairs, and walking uphill. Facial involvement is notably milder in DM2, and the characteristic hatchet face and sternocleidomastoid wasting of DM1 are typically absent or subtle. Myalgia (muscle pain) is more prominent and disabling in DM2 than in DM1, and may be the predominant complaint — some DM2 patients are initially misdiagnosed with fibromyalgia or inflammatory myopathy because pain precedes obvious weakness by years.
Back to Table of ContentsCardiac Complications
Cardiac disease is the leading cause of death in DM1, along with respiratory failure — together accounting for roughly two-thirds of disease-related mortality. The cardiac manifestations in DM1 are primarily conduction system abnormalities and arrhythmias, with cardiomyopathy as an additional but less universal feature. Understanding and actively monitoring cardiac involvement is one of the most urgent clinical priorities in DM1 management.
Progressive conduction disease begins with first-degree AV block (prolonged PR interval on ECG), which may be present even in young, mildly symptomatic patients. The conduction abnormality progresses over time to bundle branch block (left or right), bifascicular block, and ultimately complete heart block. The His-Purkinje system is particularly vulnerable, and the HV interval measured on intracardiac electrophysiology (EP) study has been established as an important risk stratifier. An HV interval greater than 70 milliseconds indicates high risk for progression to complete heart block and sudden cardiac death, and prophylactic pacemaker implantation is recommended in such patients.
Ventricular tachyarrhythmias are a separate risk — ventricular tachycardia and ventricular fibrillation can occur even in patients with seemingly modest conduction abnormalities, making the decision about implantable cardioverter-defibrillator (ICD) versus pacemaker a nuanced clinical judgment. In patients with both significant conduction disease and ventricular arrhythmia risk, a combined ICD with pacing capability is often chosen. Sudden cardiac death has been documented in DM1 patients who had only mildly abnormal baseline ECGs, underscoring the importance of longitudinal surveillance rather than a single normal baseline evaluation.
Dilated cardiomyopathy occurs in approximately 20% of DM1 patients and may be present even in patients without overt conduction disease. Systolic dysfunction can be clinically silent and detected only by echocardiography. All DM1 patients should have a 12-lead ECG at diagnosis and at least annually thereafter. Holter monitoring adds information about intermittent arrhythmias in symptomatic patients or those with AV block on resting ECG. A low threshold for cardiology referral and EP study is appropriate when any conduction abnormality is detected. DM2 patients have cardiac involvement as well, though generally milder — conduction abnormalities occur but are less common and less severe than in DM1, and sudden cardiac death risk is lower.
Back to Table of ContentsRespiratory and Sleep Involvement
Respiratory muscle weakness is the second major cause of death in DM1 and requires systematic surveillance from the time of diagnosis. The diaphragm is the primary respiratory muscle, and diaphragmatic weakness — combined with intercostal muscle weakness — reduces the capacity to generate adequate ventilatory force. This leads to progressive respiratory insufficiency, most pronounced during sleep and during intercurrent respiratory illnesses. Patients may not perceive dyspnea until respiratory reserve is severely compromised, because activity limitation from limb weakness often self-limits exertion before breathlessness becomes apparent.
Pulmonary function testing (PFTs) with forced vital capacity (FVC) is essential at baseline and at regular intervals. An FVC below 50% of predicted is associated with high risk for respiratory complications, particularly under anesthesia. Patients with severely reduced FVC may require nocturnal or even continuous bilevel positive airway pressure (BiPAP) to support ventilation. Respiratory physiotherapy and breathing exercises may help maintain chest wall compliance, and patients should receive annual influenza vaccination and pneumococcal vaccination given the aspiration and hypoventilation risk.
Sleep-disordered breathing is extremely common in DM1, affecting the majority of patients and causing one of the most disabling non-neuromuscular symptoms: hypersomnia. Sleep disturbance in DM1 takes two distinct forms. Obstructive sleep apnea arises from weakness and myotonia of pharyngeal and upper airway muscles, which collapse during sleep and obstruct airflow — the same mechanism as in primary OSA. Central sleep apnea arises from CNS dysregulation of respiratory drive (involving brainstem and thalamic dysfunction from the spliceopathy), generating irregular breathing independent of airway obstruction. Both types may coexist in the same patient and polysomnography (full overnight sleep study) is the definitive diagnostic test. CPAP addresses obstructive events; BiPAP or adaptive servo-ventilation may be needed for central or mixed patterns.
Anesthetic risk in myotonic dystrophy is one of the most practically dangerous aspects of the disease, and patients must understand and communicate this risk proactively before any procedure. Succinylcholine is absolutely contraindicated — this depolarizing muscle relaxant triggers severe, generalized myotonic spasms and potentially life-threatening hyperkalemia in DM1 patients. Non-depolarizing neuromuscular blocking agents are used with caution and with careful dosing because of prolonged effect and impaired reversal. DM1 patients have heightened sensitivity to opioids and benzodiazepines — standard doses can cause prolonged respiratory depression and apnea requiring mechanical ventilation. Regional anesthesia is preferred whenever feasible. Every DM1 patient should carry a medical alert card or bracelet identifying their diagnosis and documenting the anesthetic restrictions, and their neurologist should ideally communicate directly with the anesthesiologist before elective procedures.
Back to Table of ContentsBrain, Cognition, and Fatigue
Myotonic dystrophy is not a disease of muscle alone — the brain is substantially involved in DM1, and central nervous system symptoms are among the most disabling aspects of the condition for many patients. The same CUG expansion that disrupts splicing in muscle also affects brain tissue, where MBNL proteins are highly expressed in neurons. Brain MRI in DM1 patients typically shows white matter hyperintensities, particularly in periventricular and subcortical regions, and these correlate with cognitive and behavioral symptoms.
Hypersomnia — excessive daytime sleepiness — affects more than 70% of DM1 patients and is often described as one of the most disabling non-muscle symptoms. The sleepiness is not fully explained by nocturnal sleep disruption from sleep apnea, though co-existing sleep apnea certainly compounds it. There is evidence for a primary CNS hypersomnia related to dysfunction of brainstem and thalamic arousal circuits, analogous in some ways to narcolepsy. Patients describe irresistible sleep attacks, inability to stay awake during conversations or meals, and profound fatigue that is qualitatively different from normal tiredness. Modafinil (100–200 mg daily) is the primary pharmacological treatment and provides meaningful benefit for many patients, though it does not fully normalize alertness in severe cases.
Cognitive features in DM1 include executive dysfunction (difficulty with planning, shifting attention, and self-monitoring), visuospatial processing deficits, and reduced processing speed. Verbal intelligence and declarative memory are relatively preserved in most adult-onset DM1, but frontal-executive tasks on neuropsychological testing are typically impaired. Personality features often noted in DM1 include avoidant tendencies, rigidity, reduced initiative, and irritability — features that are partly neurological (frontal-subcortical circuit dysfunction) and partly a response to living with a debilitating progressive disease. Depression and anxiety are significantly more common in DM1 than in the general population and should be screened for and treated.
In congenital DM1 — arising from very large maternal CTG expansions — intellectual disability is a core feature, ranging from mild to severe. Children with congenital DM1 have hypotonia at birth (floppy infant), respiratory distress requiring ventilatory support, feeding difficulties, club foot (talipes), and delayed motor and cognitive milestones. Many survive to adulthood but with persistent intellectual disability. DM2 is notably milder in its CNS involvement — cognitive symptoms occur but are generally less prominent than in DM1, and there is no congenital form of DM2.
Back to Table of ContentsMultisystem: Eye, Endocrine, GI
Cataracts are one of the most characteristic and diagnostically useful extramuscular features of myotonic dystrophy. They are not ordinary cataracts but a specific type called posterior subcapsular iridescent or "Christmas tree" cataracts — multicolored, crystalline deposits in the posterior lens capsule that are visible on slit-lamp examination as a shimmering, jewel-like pattern. These cataracts can appear in the twenties or thirties, often before patients develop significant neuromuscular symptoms, and their presence in a young adult should prompt consideration of myotonic dystrophy. Routine ophthalmological examination with slit-lamp is indicated at diagnosis and should include specific documentation of posterior subcapsular change. Visual impairment from cataracts responds well to standard cataract extraction, though patients undergoing surgery face the anesthetic risks described above.
Endocrine dysfunction is multifaceted in DM1. Insulin resistance and type 2 diabetes mellitus occur at significantly higher rates than in the general population, driven in part by mis-splicing of the insulin receptor that shifts its isoform ratio toward the less-signaling IR-A form in insulin-responsive tissues. Annual fasting glucose and HbA1c screening is appropriate. Testicular dysfunction — including testicular atrophy and reduced testosterone — affects a substantial proportion of men with DM1, contributing to infertility and requiring evaluation if fathering children is desired. Thyroid dysfunction (both hypo- and hyperthyroidism) occurs at increased frequency and should be checked periodically. In women, DM1 is associated with an increased rate of miscarriage and pregnancy complications, and obstetric management requires close collaboration between neurology and maternal-fetal medicine.
Gastrointestinal involvement affects the GI tract from mouth to rectum. Dysphagia arising from pharyngeal and esophageal muscle involvement (both weakness and myotonia) is a common early symptom — patients describe difficulty initiating swallowing, frequent coughing or choking, and a sense of food sticking in the throat. Gastric dysmotility (gastroparesis) produces early satiety, nausea, and postprandial bloating. Small and large bowel dysmotility cause constipation, which can be severe and occasionally manifests as pseudo-obstruction (Ogilvie syndrome) — abdominal distension mimicking mechanical obstruction without a structural cause. Patients should be evaluated by gastroenterology if GI symptoms are prominent, and dietary modifications (small frequent meals, increased fiber, adequate hydration) are first-line management before pharmacological motility agents are considered.
Back to Table of ContentsDM1 vs DM2 Comparison
Although both DM1 and DM2 share the same basic pathogenic mechanism — RNA-mediated spliceopathy — they are sufficiently distinct clinically that careful comparison is essential for correct diagnosis and management. DM2 is sometimes called "proximal myotonic myopathy" (PROMM) to reflect its characteristic proximal weakness, and before the DM2 gene was identified in 2001 it was considered a separate entity.
| Feature | DM1 (Steinert's disease) | DM2 (Proximal Myotonic Myopathy) |
|---|---|---|
| Gene / Repeat | DMPK / CTG trinucleotide | CNBP / CCTG tetranucleotide |
| Chromosome | 19q13 | 3q21 |
| Repeat size (affected) | 50–>1,000 CTG | >75 CCTG (often thousands) |
| Anticipation | Yes, marked; maternal > paternal | Minimal / not clinically significant |
| Congenital form | Yes (maternal, >1,000 CTG) | No |
| Weakness pattern | Distal > proximal | Proximal > distal |
| Facial involvement | Prominent (ptosis, hatchet face) | Mild or absent |
| Myotonia | Grip and percussion myotonia; prominent | Present but often milder; fluctuating |
| Cardiac | Significant; common cause of death | Present but less severe |
| Cognitive / CNS | Prominent: hypersomnia, executive dysfunction | Milder; hypersomnia less common |
| Myalgia (muscle pain) | Uncommon | Prominent; often the main complaint early |
| Overall prognosis | Variable; significant morbidity and mortality | Milder; normal or near-normal lifespan common |
The proximal weakness and prominent myalgia of DM2 frequently leads to diagnostic odysseys — patients may be told they have fibromyalgia, inflammatory myopathy, or limb-girdle muscular dystrophy before the correct diagnosis is reached. Genetic testing for the DM2 CCTG expansion is technically more challenging than DM1 testing because the large repeat size makes Southern blot interpretation difficult, and specialized laboratories are needed for reliable results.
Back to Table of ContentsDiagnosis
The diagnosis of myotonic dystrophy combines clinical history, family history, examination findings, electrodiagnostic studies, and definitive genetic testing. A complete family history is essential because the autosomal dominant inheritance pattern means first-degree relatives have a 50% risk — and a parent's disease may have been mild or unrecognized. The typical clinical scenario for DM1 is a young adult presenting with hand stiffness and difficulty releasing grip, bilateral ptosis, temporal wasting, facial weakness, and mild distal limb weakness. A family history of early cataracts, diabetes, or "weakness" in relatives is an important clue.
Needle EMG is an essential early diagnostic step. Myotonic discharges — recognized by the characteristic waxing-and-waning "dive-bomber" pattern on audio — are present even in clinically asymptomatic muscles and are highly sensitive for myotonic disorders. Importantly, myotonic discharges occur in both DM1 and DM2 and cannot distinguish between them, but their presence narrows the differential markedly. Motor nerve conduction velocities are usually normal or mildly reduced, consistent with primary muscle disease. CK (creatine kinase) is elevated in most patients, typically 2- to 10-fold above normal — a modest elevation compared to Duchenne or polymyositis.
A 12-lead ECG is mandatory at the time of diagnosis — not for diagnostic purposes per se, but to identify any pre-existing conduction system disease that requires immediate cardiology evaluation. An ophthalmological examination with slit-lamp looking for posterior subcapsular cataracts is also part of the initial assessment. Pulmonary function tests (spirometry with FVC, FEV1, and maximal inspiratory/expiratory pressures) quantify respiratory involvement. A sleep study (polysomnography) should be performed if hypersomnia is present, as it will identify the type of sleep-disordered breathing and guide therapy.
Genetic testing is definitive and is required to confirm the diagnosis, determine the repeat size (which correlates roughly with severity and informs prognostication and reproductive counseling), and distinguish DM1 from DM2. For DM1, PCR amplification followed by Southern blot hybridization (for large expansions beyond the range of standard PCR) quantifies the CTG repeat length. For DM2, repeat-primed PCR and Southern blot of the CCTG expansion in CNBP is used. Muscle biopsy is rarely needed if genetic testing is available — when performed, it shows type 1 fiber atrophy, increased central nuclei, ring fibers, and sarcoplasmic masses, findings characteristic but not pathognomonic. Molecular testing has largely supplanted biopsy for diagnostic confirmation.
Back to Table of ContentsTreatment and Management
There is currently no approved disease-modifying therapy that slows the underlying progression of myotonic dystrophy, but multiple investigational approaches targeting the RNA pathology are in clinical trials. Antisense oligonucleotides (ASOs) designed to degrade the toxic CUG RNA or to block its binding to MBNL proteins have shown efficacy in DM1 mouse models and are advancing toward human trials. Small molecules that disrupt the CUG RNA hairpin structure and prevent MBNL sequestration are another active research area. Gene silencing approaches targeting the expanded DMPK allele specifically are also under development. Patients should be encouraged to enroll in clinical trials when eligible, and resources such as the Myotonic Dystrophy Foundation (MDF) or ClinicalTrials.gov can help identify opportunities.
Mexiletine is the most important pharmacological treatment currently available for symptomatic myotonia. It is a class Ib sodium channel blocker (originally an antiarrhythmic) that reduces membrane hyperexcitability by blocking the persistent sodium currents that sustain repetitive action potentials. At doses of 150–200 mg three times daily, mexiletine significantly reduces grip myotonia and percussion myotonia in DM1 patients. A pivotal randomized controlled trial (Logigian et al., 2010) demonstrated its efficacy. Mexiletine is generally well tolerated, with the main concerns being GI side effects (nausea, taken with food) and a small pro-arrhythmic risk, making a baseline ECG and cardiology sign-off appropriate before initiation. Patients should be counseled that mexiletine treats stiffness (myotonia) but does not improve weakness.
Cardiac management is guided by degree of conduction abnormality. Cardiologists experienced with neuromuscular disease should participate in care. Guidelines recommend pacemaker implantation for patients with complete heart block, symptomatic bradycardia, or significant HV prolongation on EP study (>70 ms). ICD is added for patients with ventricular arrhythmia risk. Annual ECG surveillance is standard; Holter monitoring adds sensitivity for intermittent arrhythmias. Patients should understand that a normal annual ECG does not guarantee safety — sudden cardiac death can occur between monitoring intervals, and awareness of syncope, palpitations, or near-syncope as warning symptoms is important.
Respiratory management includes pulmonary function testing at baseline and every 1–2 years. BiPAP titration during polysomnography is appropriate for those with significant nocturnal hypoventilation or sleep apnea. Pulmonary rehabilitation improves exercise tolerance and may help maintain respiratory muscle function. Annual influenza and pneumococcal vaccination are essential given the aspiration and hypoventilation risk. Physical therapy prevents contractures, maintains strength in preserved muscle groups, and fits orthotic devices such as ankle-foot orthoses (AFOs) for foot drop. Occupational therapy addresses adaptive equipment needs for daily living activities.
For hypersomnia, modafinil (200 mg in the morning, sometimes with an additional 100 mg at noon) is first-line and has demonstrated benefit in trials. Methylphenidate and sodium oxybate have also been used. Cognitive and psychiatric symptoms — depression, anxiety, executive dysfunction — are managed with standard pharmacological and psychological approaches, noting that DM1 patients may have heightened sensitivity to psychotropic medications. Regular genetic counseling for the patient and family members is an ongoing need, particularly for reproductive decisions. MedicAlert identification is non-negotiable for all DM1 patients given the anesthetic risks. Comprehensive care through a multidisciplinary neuromuscular clinic — neurologist, cardiologist, pulmonologist, physiotherapist, genetic counselor — yields the best outcomes.
Back to Table of ContentsKey Research Papers
- Brook JD et al. (1992) — Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell. 68:799-808. — Search PubMed
- Liquori CL et al. (2001) — Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 293:864-7. PMID 11486088
- Miller JW et al. (2000) — Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 19:4439-48. — Search PubMed
- Mankodi A et al. (2002) — Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 10:35-44. — Search PubMed
- Day JW et al. (2003) — Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 60:657-64. — Search PubMed
- Ranum LP & Day JW (2004) — Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet. 74:793-804. — Search PubMed
- Groh WJ et al. (2008) — Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. NEJM. 358:2688-97. — Search PubMed
- Udd B & Krahe R (2012) — The myotonic dystrophies: molecular, clinical and therapeutic challenges. Lancet Neurol. 11:891-905. — Search PubMed
- Wahbi K et al. (2012) — Electrophysiological study with prophylactic pacing and survival in adults with myotonic dystrophy and conduction system disease. JAMA. 307:1292-301. — Search PubMed
- Logigian EL et al. (2010) — Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology. 74:1441-8. — Search PubMed
- Overend G et al. (2014) — Splice isoform switching and mis-splicing in myotonic dystrophy. Skelet Muscle. 4:8. — Search PubMed
- Meola G & Cardani R (2015) — Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta. 1852:594-606. — Search PubMed
Connections
- Neurology
- Lambert-Eaton Syndrome
- Myasthenia Gravis
- Charcot-Marie-Tooth
- Multiple Sclerosis
- Guillain-Barré Syndrome
- Diseases