Duchenne Muscular Dystrophy (DMD)
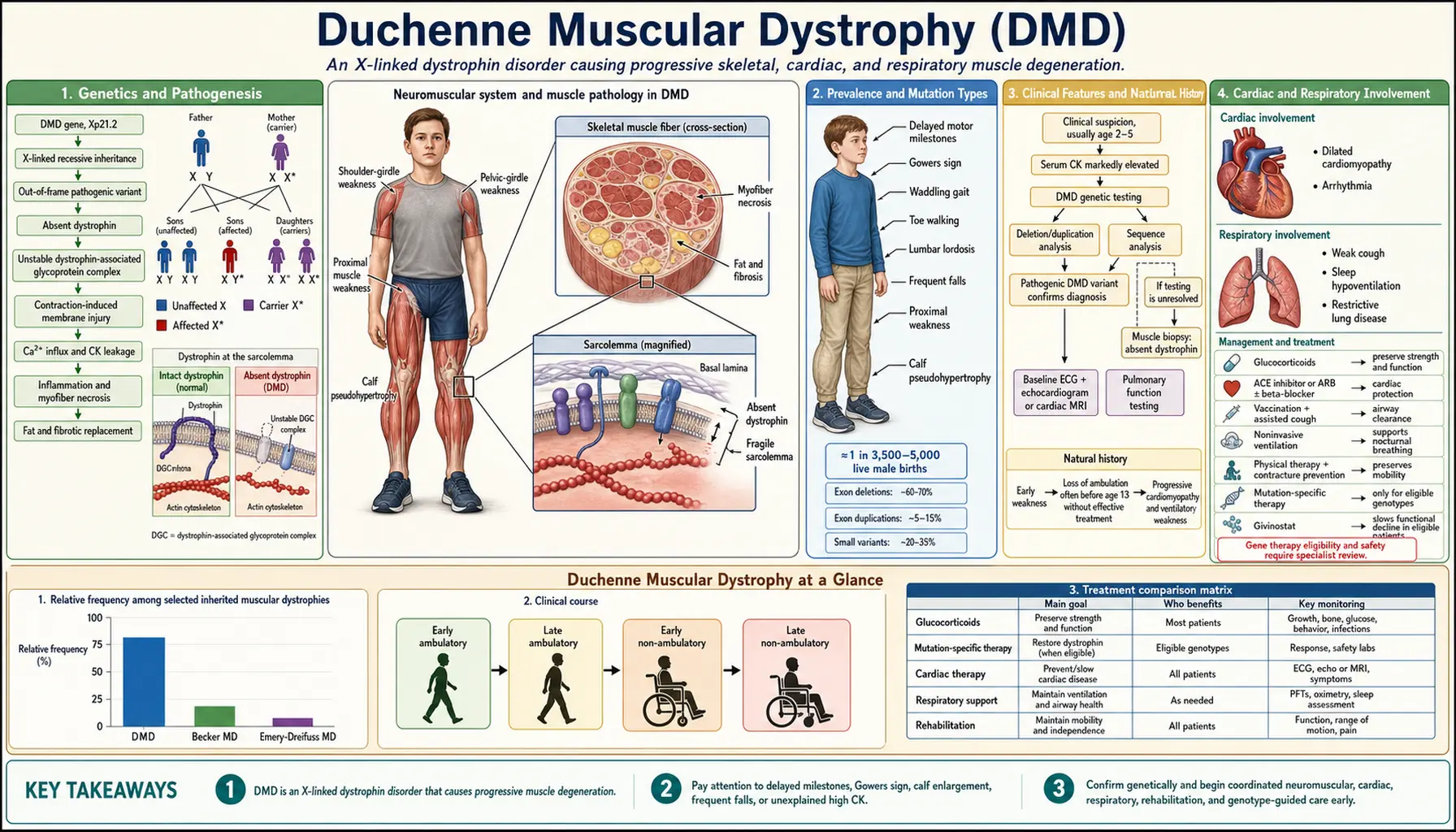
DMD is an X-linked recessive neuromuscular disease caused by out-of-frame mutations in the DMD gene (Xp21.2), the largest gene in the human genome at 2.4 megabases, resulting in complete absence of dystrophin protein. Without dystrophin anchoring the dystrophin-glycoprotein complex (DGC) to the sarcolemma, repeated contraction cycles cause membrane micro-tears, calcium influx, fiber necrosis, and progressive replacement by fibrofatty connective tissue. DMD affects approximately 1 in 3,500 live male births and is uniformly progressive — but modern gene-based therapies have transformed the therapeutic landscape.
Interactive Visualization Muscle Contraction — release the calcium Expose the actin binding sites and watch myosin heads ratchet through the cross-bridge cycle to shorten the sarcomere. Launch →Table of Contents
- Genetics and Pathogenesis
- Prevalence and Mutation Types
- Clinical Features and Natural History
- Cardiac and Respiratory Involvement
- Diagnosis
- Corticosteroid Therapy
- Gene-Based and Molecular Therapies
- Key Research Papers
- Featured Videos
1. Genetics and Pathogenesis
The DMD gene resides at chromosomal locus Xp21.2 and spans 2.4 megabases across 79 exons, making it the largest gene in the human genome. Its sheer size partly explains its high spontaneous mutation rate — approximately one-third of affected boys carry a new, de novo mutation with no family history of the disease.
The Reading Frame Rule governs clinical severity. Out-of-frame mutations disrupt the translational reading frame downstream of the deletion or insertion, introducing a premature stop codon and preventing production of any functional dystrophin protein — this is Duchenne MD. In-frame mutations preserve the reading frame and allow synthesis of a shortened but partially functional protein — this is the milder Becker MD. This rule correctly predicts phenotype in roughly 90% of patients and is the mechanistic rationale behind exon-skipping therapies.
Dystrophin functions as a molecular shock-absorber, linking the intracellular actin cytoskeleton to the extracellular matrix through the dystrophin-glycoprotein complex (DGC). The DGC includes sarcoglycans, dystroglycans, syntrophins, and dystrobrevins. Loss of dystrophin destabilizes the entire complex, rendering the sarcolemma mechanically fragile. During repeated contraction-relaxation cycles, micro-tears develop, allowing pathological calcium influx. Elevated intracellular calcium activates calpain proteases and triggers mitochondrial dysfunction, leading to myofiber necrosis. Inflammatory infiltrates (macrophages, CD8+ T cells) amplify damage, and the regenerative capacity of muscle stem cells (satellite cells) is eventually exhausted, leaving fibrofatty connective tissue in place of contractile muscle.
Brain-specific dystrophin isoforms explain the non-motor features of DMD. The Dp140 isoform is expressed in hippocampal neurons and is absent in patients with mutations downstream of exon 45, correlating with greater cognitive and behavioral impact. The Dp71 isoform is expressed throughout the brain and its loss contributes to intellectual disability, attention deficits, and autism spectrum features seen in 30–40% of boys with DMD.
2. Prevalence and Mutation Types
DMD occurs in approximately 1 in 3,500 live male births worldwide, translating to roughly 20,000 new cases per year globally and an estimated 15,000 patients currently living with DMD in the United States. The disease is pan-ethnic with no significant variation by ancestry.
Large intragenic deletions account for 65–70% of DMD mutations, with two hotspot regions: exons 2–20 and exons 45–55. Duplications account for another 10–15% of cases. Point mutations make up the remainder — nonsense (premature stop) mutations represent approximately 10%, missense mutations approximately 5%, and intronic or splice-site changes are rare. This distribution is clinically important because each mutation class is amenable to different therapeutic strategies.
Approximately one-third of cases arise from de novo mutations, meaning neither parent carries the variant and genetic counseling must account for the possibility of gonadal mosaicism even when parental testing is negative. Females with one mutated X chromosome are typically carriers. However, approximately 8% of manifesting carriers develop clinically significant cardiomyopathy, reflecting skewed X-inactivation that silences the normal allele in cardiac tissue — these women warrant the same cardiac surveillance protocol as affected males.
Genetic testing begins with multiplex ligation-dependent probe amplification (MLPA), which efficiently detects deletions and duplications across all 79 exons. If MLPA is uninformative, Sanger or next-generation sequencing identifies point mutations. Newborn screening programs using creatine kinase measurement on dried blood spots are expanding in the United States and several other countries, enabling pre-symptomatic diagnosis and earlier access to approved gene therapies.
3. Clinical Features and Natural History
The earliest motor signs of DMD typically emerge between ages 2 and 5, when parents notice delayed walking, frequent falls, difficulty climbing stairs, or reluctance to run. Because the weakness is proximal and symmetric, it preferentially affects hip extensors, quadriceps, and shoulder girdle muscles before distal limb muscles.
Gower's Sign is the hallmark clinical finding: when asked to rise from the floor, the child places hands on thighs and "walks" them upward to compensate for weak hip extensors and quadriceps. This maneuver is not pathognomonic for DMD but is highly characteristic when observed in a young boy with markedly elevated creatine kinase.
Calf pseudohypertrophy — enlarged calves that appear muscular but are firm and non-functional — results from fibrofatty infiltration rather than true muscle hypertrophy. It is present in the majority of boys with DMD and is a useful early diagnostic clue.
Gait abnormalities include a Trendelenburg pattern (lateral trunk sway from gluteus medius weakness), toe-walking (equinus posture from early heel-cord contracture), and a waddling quality from bilateral hip weakness. Lumbar lordosis is accentuated as the child uses spinal extension to maintain balance.
Serum creatine kinase is characteristically elevated to 10,000–50,000 IU/L, representing 10 to 50 times the upper limit of normal. Crucially, CK is elevated from birth — before any clinical weakness — because dystrophin deficiency causes continuous subclinical membrane damage even in infants. This makes CK a useful newborn screening biomarker.
Without corticosteroid treatment, most boys lose independent ambulation between ages 9 and 12. Corticosteroids extend the ambulatory period by 2–5 years. Once wheelchair-dependent, secondary complications accelerate: scoliosis develops in up to 90% of untreated non-ambulatory patients (reduced to approximately 20% with corticosteroids), joint contractures progress at the hip flexors, heel cords, and iliotibial band, and osteoporosis is compounded by both the disease process and corticosteroid therapy. Fractures — particularly vertebral compression fractures — are a significant source of morbidity.
4. Cardiac and Respiratory Involvement
Cardiac involvement is universal in DMD males. Dilated cardiomyopathy develops in virtually 100% by early adulthood, and cardiac disease shares the position of leading cause of death alongside respiratory failure. The distinctive early pattern on cardiac MRI is posterolateral left ventricular free wall fibrosis — reflecting the regional distribution of mechanical stress on a dystrophin-deficient myocardium. Electrocardiography characteristically shows a tall R-wave in lead V1 and deep Q-waves in lateral leads (I, aVL, V5-V6), a pattern that reflects posterior wall fibrosis and can precede echocardiographic dysfunction by years.
Cardiac surveillance should begin at diagnosis (or by age 6 at the latest) with echocardiography and ECG, transitioning to cardiac MRI by age 10 for more sensitive fibrosis mapping. Cardiac MRI with late gadolinium enhancement quantifies the extent of myocardial fibrosis and predicts future functional decline.
Treatment of DMD cardiomyopathy follows heart failure guidelines adapted to this population. ACE inhibitors (lisinopril, enalapril) are recommended to begin by age 10 or at the first sign of dysfunction, with evidence suggesting that pre-emptive initiation in patients with normal function may delay onset of overt cardiomyopathy. ARBs are used when ACE inhibitors are not tolerated. Beta-blockers are added once systolic dysfunction is established. The EIDMD trial demonstrated a benefit for eplerenone (a mineralocorticoid receptor antagonist) in combination with ACE inhibition in delaying progression. Cardiac transplantation is considered in carefully selected patients with refractory heart failure and preserved skeletal muscle function.
Manifesting female carriers with cardiomyopathy require the same treatment approach and warrant dedicated cardiac surveillance beginning no later than their twenties.
Respiratory muscle weakness follows a predictable course after loss of ambulation. Forced vital capacity (FVC) declines at approximately 6–8% per year once the patient is non-ambulatory. Non-invasive ventilation (bilevel positive airway pressure, BiPAP) is initiated when FVC falls below 50% of predicted, when symptomatic nocturnal hypoventilation is documented on overnight oximetry or capnography, or when daytime hypercapnia appears. Cough assistance — manual-assisted cough, mechanical insufflation-exsufflation (MI-E) devices — is critical for secretion clearance and prevention of pneumonia. Annual pulmonary function testing begins at age 5–6. Invasive ventilation via tracheostomy remains an option for patients or families who choose maximum ventilatory support.
With comprehensive multidisciplinary care including corticosteroids, cardiac medications, and ventilatory support, survival commonly extends into the 30s and 40s — a dramatic improvement over the median survival of late teens seen in earlier decades.
5. Diagnosis
The diagnostic pathway typically begins with serum creatine kinase, which is markedly elevated (10,000–50,000 IU/L) in virtually all boys with DMD. Because CK is elevated from birth, it is an effective newborn screening marker; programs using CK measurement on dried blood spot cards are active in several US states and are expanding internationally.
Genetic testing has become the definitive diagnostic step. MLPA is the first-line test, capable of detecting deletions and duplications across all 79 exons with high sensitivity and specificity. When MLPA is non-diagnostic, targeted or whole-exome sequencing identifies point mutations including nonsense variants, missense changes, and splice-site alterations. Genetic confirmation identifies the precise mutation, which is critical for determining eligibility for specific exon-skipping therapies, establishing carrier status in female relatives, and guiding genetic counseling.
Muscle biopsy is increasingly reserved for cases where genetic testing is inconclusive. When performed, biopsy shows absence of dystrophin staining on immunohistochemistry and absence of the protein on Western blot. Histological findings include marked variation in fiber size, frequent necrotic and regenerating fibers, endomysial fibrosis, and fibrofatty replacement — changes that reflect the ongoing cycle of degeneration and failed regeneration.
Brain MRI may be obtained when cognitive or behavioral concerns are prominent, particularly in patients with mutations affecting the Dp140 or Dp71 isoforms. Cardiac MRI with late gadolinium enhancement provides superior characterization of myocardial fibrosis compared to echocardiography alone and is now considered standard in the cardiac workup.
The differential diagnosis includes Becker muscular dystrophy (milder course, in-frame mutations, partially functional dystrophin), limb-girdle muscular dystrophies (autosomal inheritance, sarcoglycan or calpain mutations), spinal muscular atrophy (lower motor neuron, normal CK), and congenital myopathies (structural protein mutations, often present at birth).
6. Corticosteroid Therapy
Corticosteroids remain the backbone of pharmacological management for DMD and are the only drugs with established Level I evidence for extending ambulation and slowing respiratory decline. Two agents are in common use: prednisone, available generically, and deflazacort (brand name Emflaza), which received FDA approval in 2017 specifically for DMD.
Deflazacort is dosed at 0.9 mg/kg/day and has demonstrated comparable or superior efficacy to prednisone for preserving motor function. Its principal advantage is a lower rate of weight gain compared to prednisone, a clinically significant difference given that obesity compounds ambulation difficulty and cardiac load. However, deflazacort carries a higher rate of posterior subcapsular cataracts than prednisone, requiring annual ophthalmological surveillance. The comparative data were formalized in a landmark trial by Griggs and colleagues published in 2016 (PMID: 27770074), which showed that deflazacort-treated patients were more likely to remain ambulatory at 52 weeks compared to placebo and had numerically better outcomes than prednisone, though the trial was not powered for a head-to-head superiority comparison.
Dosing strategies fall into two main categories: daily continuous dosing, which appears most efficacious for preserving ambulation; and intermittent regimens such as 10-days-on/10-days-off or weekend-only dosing, which are sometimes preferred to reduce growth suppression and behavioral side effects while maintaining some motor benefit. The Cooperative International Neuromuscular Research Group (CINRG) network and ACTION Duchenne have contributed large natural history and observational datasets that inform guideline development.
The benefits of corticosteroid therapy extend beyond ambulation. Treatment reduces the incidence of scoliosis from approximately 90% to roughly 20% in non-ambulatory patients — a benefit large enough to frequently eliminate the need for spinal surgery. Corticosteroids also moderate the rate of respiratory decline and appear to reduce the severity of cardiomyopathy onset, though cardiac benefit is less well quantified than the motor benefit.
Side effects require proactive management. Weight gain and cushingoid features are common; dietary counseling and physical activity optimization help mitigate them. Growth delay is an expected effect and should be monitored longitudinally. Behavioral changes — irritability, emotional lability, hyperactivity — are frequent and may require dose adjustment or consultation with a behavioral specialist. Adrenal suppression mandates stress-dose coverage during illness, surgery, or injury; corticosteroids must never be abruptly discontinued. Bone health is addressed with supplemental vitamin D and calcium from the start of treatment, with DXA scanning to monitor bone mineral density and prompt treatment of vertebral fractures.
7. Gene-Based and Molecular Therapies
The past decade has produced a dramatic expansion of approved molecular and gene-based therapies for DMD, driven by the reading-frame rule and advances in antisense chemistry and viral gene delivery. These therapies target the specific molecular defect rather than downstream inflammation or fibrosis.
Exon skipping with antisense oligonucleotides (ASOs) is the most broadly applicable molecular strategy. ASOs are short synthetic nucleic acid sequences that bind to pre-mRNA and induce targeted skipping of one or more exons during splicing. By excluding a specific out-of-frame exon, the technique restores the reading frame downstream of a deletion, converting a Duchenne-type out-of-frame mutation into a Becker-type in-frame mutation and enabling production of a shortened but partially functional dystrophin. Four ASO drugs have received FDA accelerated approval, each targeting a different exon and covering a distinct patient subset: eteplirsen (Exondys 51; 2016) for exon 51 skipping, applicable to approximately 13% of DMD mutations; golodirsen (Vyondys 53; 2019) for exon 53 skipping; viltolarsen (Viltepso; 2020) also targeting exon 53; and casimersen (Amondys 45; 2021) for exon 45 skipping. Collectively these four drugs are applicable to approximately 30% of DMD patients with eligible deletion mutations. Clinical confirmatory trials are ongoing.
Nonsense readthrough is the therapeutic approach for patients with premature stop codons, approximately 10% of DMD mutations. Ataluren (Translarna) induces ribosomal readthrough of premature stop codons, allowing translation to continue past the mutation and produce full-length dystrophin at reduced efficiency. Ataluren is conditionally approved in the European Union and several other countries but has not received FDA approval in the United States following review of confirmatory trial data.
Micro-dystrophin gene therapy represents a mutation-agnostic approach applicable to the full DMD population regardless of specific mutation. Delandistrogene moxeparvovec (Elevidys; Sarepta Therapeutics) uses an adeno-associated virus serotype rh74 (AAVrh74) vector to deliver a truncated micro-dystrophin transgene that retains the most mechanically critical functional domains. The FDA granted accelerated approval in June 2023 for ambulatory patients ages 4–5, based on micro-dystrophin protein expression data and early functional outcomes. Broader age range extension and full approval pathway are under active investigation, with longer-term follow-up data accumulating from the ENDEAVOR and EMBARK trials.
The pipeline extends to several additional approaches at earlier stages of development. CRISPR-based exon deletion strategies (Editas Medicine, Exonics Therapeutics, now Sarepta) aim to permanently excise out-of-frame exons from genomic DNA in muscle cells. Utrophin upregulation — inducing expression of the structurally related protein utrophin, which can partially substitute for dystrophin — has been explored with agents including ezutromid, though clinical results have been mixed. Stem cell approaches using satellite cell delivery or iPSC-derived myogenic progenitors are in pre-clinical and early clinical stages.
All patients with DMD, regardless of which specific molecular therapy they are receiving or whether they are eligible for approved therapies, benefit from comprehensive multidisciplinary care integrating neurology, cardiology, pulmonology, orthopedics, physical and occupational therapy, nutrition, psychology, and genetics. The emergence of gene-based options does not replace this coordinated model — it is layered on top of it.
8. Key Research Papers
- Mendell JR et al. "Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy." Ann Neurol. 2016;79(2):257–271. — Search PubMed
- Griggs RC et al. "Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy." Neurology. 2016;87(20):2123–2131. — Search PubMed
- Birnkrant DJ et al. "Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management." Lancet Neurol. 2018;17(3):251–267. PMID: 29395989
- Birnkrant DJ et al. "Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management." Lancet Neurol. 2018;17(4):347–361. PMID: 29395990
- Duan D et al. "Duchenne muscular dystrophy." Nat Rev Dis Primers. 2021;7(1):13. PMID: 33602943
- Mendell JR et al. "Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children with Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial." JAMA Neurol. 2020;77(9):1122–1131. PMID: 32539076
- Eagle M et al. "Managing Duchenne muscular dystrophy — the additive effect of spinal surgery and home nocturnal ventilation in improving survival." Neuromuscul Disord. 2007;17(6):470–475. — Search PubMed
- Thrush PT et al. "Re-examination of the cardiomyopathy in Duchenne muscular dystrophy." Can J Cardiol. 2009;25(11):e410–414. — Search PubMed
- Mavrogeni S et al. "Cardiac and skeletal muscle MRI for evaluation of cardiomyopathy in Duchenne muscular dystrophy." JACC Cardiovasc Imaging. 2015;8(12):1444–1456. — Search PubMed
- Bladen CL et al. "The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations." Hum Mutat. 2015;36(4):395–402. PMID: 25604253
- Matthews E et al. "Corticosteroids for the treatment of Duchenne muscular dystrophy." Cochrane Database Syst Rev. 2016;(5):CD003725. PMID: 27149418
- Hoffman EP et al. "Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy." N Engl J Med. 1988;318(21):1363–1368. — Search PubMed
Connections
- Neurology
- Muscle Contraction — interactive animation
- Becker Muscular Dystrophy
- Facioscapulohumeral Muscular Dystrophy (FSHD)
- Myotonic Dystrophy
- Spinal Muscular Atrophy
- ALS
- Myasthenia Gravis
- Charcot-Marie-Tooth Disease
- Lab Tests