N-acetyl-cysteine (NAC) as the Acetaminophen (Paracetamol) Antidote
N-acetylcysteine is the only proven antidote for acetaminophen (paracetamol) overdose — the single most common cause of acute liver failure in the United States and the United Kingdom. When given within 8 hours of ingestion it delivers essentially 100% transplant-free survival. After 8 hours efficacy declines steeply, and beyond 24 hours the role becomes supportive rather than preventive. This deep-dive walks through the NAPQI/CYP2E1 mechanism, the two standard dosing protocols (Prescott IV and Smilkstein oral), the Rumack-Matthew nomogram, the special populations that change the calculus, and why no other intervention — not activated charcoal, not methionine, not silymarin — matches NAC's clinical efficacy in this indication.
Interactive Visualization Free Radicals & Your Antioxidant Network Follow one superoxide radical down the whole relay — SOD, catalase, glutathione, vitamin E, vitamin C, NADPH — then release free iron and watch a membrane tear itself apart. Launch →
Table of Contents
- Why Acetaminophen Is Dangerous
- NAPQI: The Toxic Intermediate
- CYP2E1 — the Enzyme That Makes the Poison
- Glutathione Depletion — the Tipping Point
- How NAC Reverses the Toxicity
- The Rumack-Matthew Nomogram
- Prescott IV Protocol (21-Hour)
- Smilkstein Oral Protocol (72-Hour)
- The 8-Hour Window — Why Timing Is Everything
- Special Populations and Risk Modifiers
- Adverse Reactions to IV NAC
- Why NAC Is the Only Proven Antidote
- Cautions
- Key Research Papers
- Connections
- Featured Videos
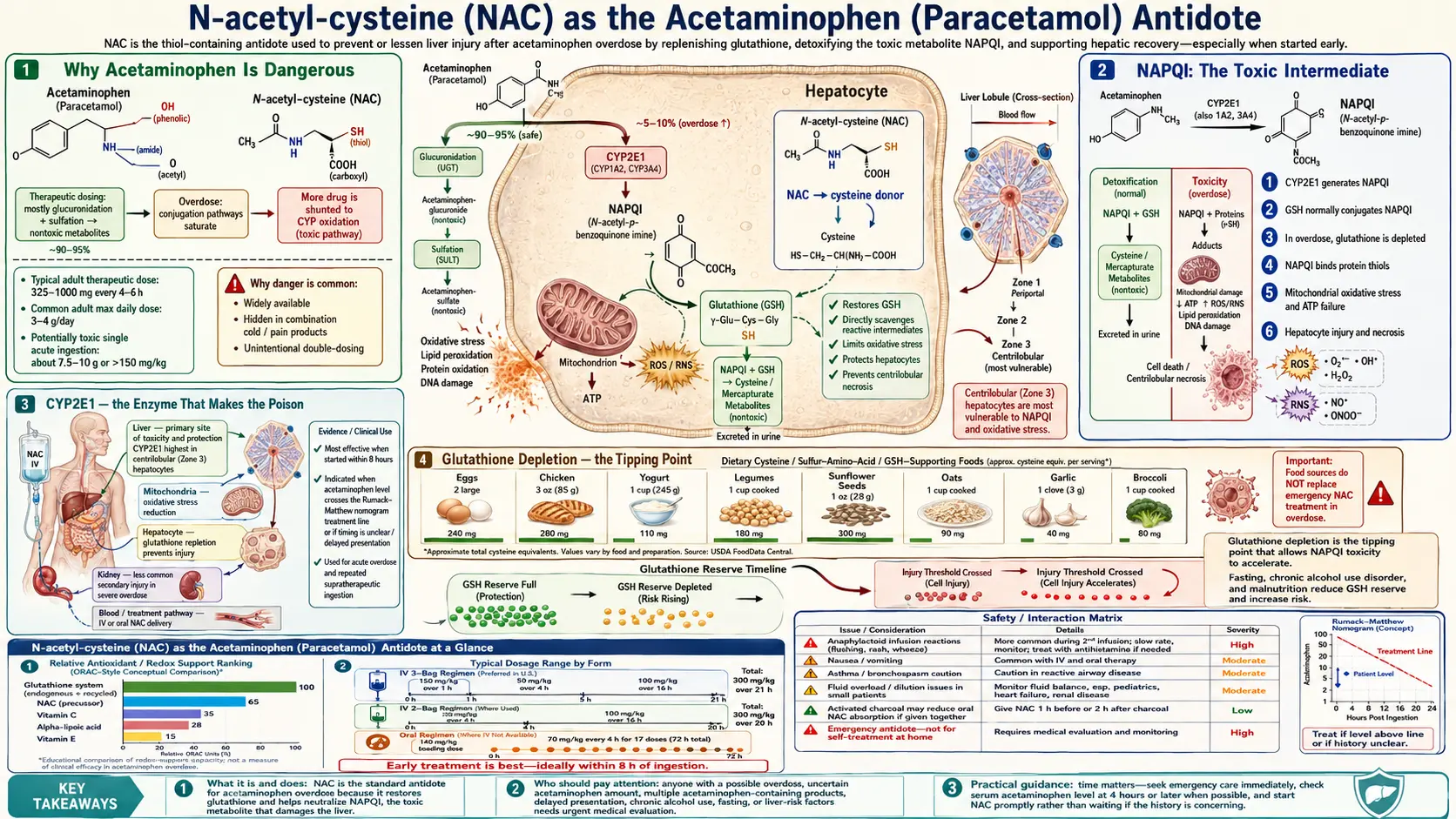
Why Acetaminophen Is Dangerous
Acetaminophen (US name) and paracetamol (rest of the world) is the most widely used over-the-counter analgesic in the world. At therapeutic doses (up to 4 g/day in adults, less in children and people with chronic alcohol use) it is one of the safer pain relievers in routine clinical use. The danger appears with overdose — whether intentional (a common method of self-harm) or unintentional (often from combining multiple acetaminophen-containing products such as Tylenol plus Vicodin plus a cold medicine).
Acetaminophen-induced acute liver failure (ALF) is now the single most common cause of ALF in the US, UK, and most of Western Europe, accounting for approximately 40–50% of cases. Roughly 50,000 emergency-department visits and 500 deaths per year in the US are attributable to acetaminophen overdose. The toxicity is dose-dependent and predictable, which is exactly why NAC works: the clock starts at the moment of ingestion, and the chemistry of what happens next is well understood.
The toxic threshold in healthy adults is approximately 150 mg/kg or 7.5 g, whichever is lower — though some patients show injury at lower exposures, particularly with chronic alcohol use, malnutrition, or prolonged fasting. Doses above 250 mg/kg are essentially always toxic without intervention.
NAPQI: The Toxic Intermediate
At therapeutic doses about 90% of acetaminophen is metabolized through safe Phase II conjugation — glucuronidation (about 50–60%) and sulfation (about 25–35%) — both producing water-soluble metabolites that are excreted in urine. A small fraction (about 5–10%) is oxidized by hepatic cytochrome P450 enzymes (primarily CYP2E1, with minor contributions from CYP1A2 and CYP3A4) into a highly reactive electrophilic intermediate: N-acetyl-p-benzoquinone imine (NAPQI).
At therapeutic doses, NAPQI is immediately neutralized by conjugation with glutathione, forming a mercapturic acid that is excreted in urine. The hepatocyte's glutathione pool handles the load comfortably. In overdose, three things change simultaneously:
- Glucuronidation and sulfation saturate. The Phase II enzymes work near maximum capacity. A larger fraction of incoming acetaminophen is shunted toward CYP2E1.
- NAPQI production accelerates. More substrate flows into the oxidative pathway. NAPQI concentration in the hepatocyte rises.
- Glutathione is consumed. Each NAPQI molecule requires one glutathione molecule to neutralize. The hepatic GSH pool is finite (about 5–10 mmol/kg in a healthy liver) and is depleted within hours.
Once glutathione drops below approximately 30% of baseline, NAPQI accumulates and begins binding to hepatocyte proteins — particularly mitochondrial proteins — through covalent adduct formation with cysteine residues. The downstream consequences are mitochondrial dysfunction, ATP depletion, mitochondrial permeability transition pore opening, oxidative stress, and centrilobular hepatocyte necrosis. The classic histology — zone 3 (centrilobular) necrosis with intact periportal hepatocytes — reflects the centrilobular concentration of CYP2E1.
CYP2E1 — the Enzyme That Makes the Poison
CYP2E1 is the primary cytochrome P450 responsible for generating NAPQI. Its expression and activity vary substantially among patients, which explains much of the inter-individual variability in acetaminophen toxicity. Three classes of CYP2E1 induction are clinically important:
- Chronic alcohol use — ethanol both induces CYP2E1 (more enzyme is made) and depletes hepatic glutathione (less defense available). The combination doubles or triples acetaminophen hepatotoxicity risk at any given dose. The classic "therapeutic misadventure" is a chronic drinker who takes 4–6 g/day of acetaminophen for hangover headaches and develops ALF at a dose that would be perfectly safe for a non-drinker.
- Isoniazid (and other CYP2E1 inducers) — isoniazid for tuberculosis prophylaxis is a well-documented potentiator of acetaminophen hepatotoxicity. Chronic ketogenic diets also induce CYP2E1 because ketone bodies are substrates.
- Fasting and malnutrition — depletes the hepatic glucuronide pool (less Phase II conjugation capacity) and depletes glutathione (less NAPQI neutralization). Patients who present after a multi-day binge without eating, or chronically malnourished alcoholics, are at substantially elevated risk.
Acute alcohol intake (a single binge) actually protects against acetaminophen toxicity because ethanol competes with acetaminophen for CYP2E1 binding sites. Only chronic alcohol use is the risk amplifier.
Glutathione Depletion — the Tipping Point
The entire pathophysiology of acetaminophen toxicity can be summarized in one sentence: NAPQI is harmless if glutathione is available to neutralize it, and lethal if glutathione runs out. The clinical task is therefore exactly equivalent to the biochemical task — keep hepatocyte glutathione above the threshold at which NAPQI starts adducting to mitochondrial proteins.
Glutathione is a tripeptide of glutamate, cysteine, and glycine. The rate-limiting step in its synthesis is the action of glutamate-cysteine ligase (GCL) on these substrates, and the rate-limiting substrate is cysteine. Free cysteine cannot be reliably given orally — it is largely degraded by gut bacteria and rapidly oxidized in the bloodstream to cystine (the disulfide), which the hepatocyte cannot use directly. This is the problem that NAC solves — see the Glutathione Precursor deep-dive for the full pharmacokinetic story.
The 8-hour clinical window for NAC reflects this glutathione kinetics. In the first 4 hours after a toxic ingestion, acetaminophen is still being absorbed and the glutathione pool is being drained but not yet depleted. From 4 to 8 hours, glutathione approaches the critical threshold. Beyond 8 hours, mitochondrial protein adducts have formed; even if glutathione is restored, the structural damage to hepatocyte mitochondria is already in motion and progresses through the next 24–72 hours regardless of whether further NAPQI is being made.
How NAC Reverses the Toxicity
NAC works through four overlapping mechanisms when given for acetaminophen toxicity, in roughly this order of importance:
- Glutathione precursor — the dominant mechanism. NAC is deacetylated to cysteine, cysteine enters the GCL reaction, and hepatic glutathione is restored. Tissue GSH begins rising within hours of starting NAC and reaches normal range within 24 hours of consistent dosing.
- Direct NAPQI conjugation — the free thiol on NAC can react directly with NAPQI to form a stable adduct, neutralizing the toxin before it reaches mitochondrial proteins. This is most relevant in the first 4–8 hours when NAPQI generation is peaking.
- Sulfation substrate — NAC also provides sulfate (via cysteine catabolism) that can shift residual acetaminophen toward the non-toxic Phase II sulfation pathway, away from CYP2E1.
- Mitochondrial support — beyond the 8-hour window, when NAPQI adducts are already present, NAC provides direct antioxidant support to mitochondria, may reduce inflammatory cytokine output through NF-κB suppression, and supports microcirculation through nitric oxide modulation. This is why the Lee 2009 trial showed survival benefit even when NAC was started after acetaminophen-independent acute liver failure was already in motion.
The Rumack-Matthew Nomogram
Published in Pediatrics in 1975 by Barry Rumack and Henry Matthew, this nomogram remains the gold-standard tool for risk stratification after acute single-time-point acetaminophen ingestion. The nomogram plots serum acetaminophen concentration on a log scale against time since ingestion on a linear scale. Two parallel diagonal lines descend from upper-left to lower-right:
- The "probable hepatotoxicity" line — originally drawn at 200 mcg/mL at 4 hours. Patients with a 4-hour level at or above this line have a high probability of hepatic injury without treatment.
- The "possible hepatotoxicity" line — drawn at 150 mcg/mL at 4 hours (the current US treatment threshold). Patients at or above this lower line are treated empirically with NAC even though some would not develop injury.
The clinical workflow is straightforward: measure serum acetaminophen at 4 hours post-ingestion (or as soon as possible after that), plot the result on the nomogram, and start NAC if the level is at or above the 150 mcg/mL line. Levels drawn before 4 hours are not interpretable on the nomogram because absorption is incomplete.
The nomogram does not apply to:
- Staggered or repeated supratherapeutic ingestions over hours or days (no single time-zero)
- Unknown time of ingestion
- Extended-release or modified-release acetaminophen formulations
- Patients presenting more than 24 hours after ingestion
In these settings, the decision to treat is made on the basis of acetaminophen level (any detectable level), elevated transaminases, or clinical concern. The default for any uncertain case is to treat empirically — the risk-benefit calculus strongly favors unnecessary NAC over missed hepatotoxicity.
Prescott IV Protocol (21-Hour)
The intravenous protocol was originally published by Laurie Prescott in Edinburgh in 1979. It has been the standard worldwide IV regimen for over four decades and remains essentially unchanged. The total NAC dose is 300 mg/kg, delivered in three sequential infusions over 21 hours:
| Bag | Dose | Duration | Diluent (typical) |
|---|---|---|---|
| Loading | 150 mg/kg | 60 minutes (originally 15) | 200 mL D5W |
| Maintenance 1 | 50 mg/kg | 4 hours | 500 mL D5W |
| Maintenance 2 | 100 mg/kg | 16 hours | 1000 mL D5W |
The original Prescott protocol called for the loading dose to run over 15 minutes; this has been extended to 60 minutes in most modern guidelines to reduce the rate of anaphylactoid reactions, which is dose-rate-dependent. The total elapsed time is 21 hours, which is why the regimen is often called simply "the 21-hour protocol."
At the end of the 21-hour infusion, the patient is reassessed. If transaminases are rising, INR is elevated, or acetaminophen is still detectable, the maintenance infusion is continued (typically at 6.25 mg/kg/h) until the patient is clearly improving. The SNAP trial (Bateman 2014) tested a shortened 12-hour regimen with comparable efficacy and lower anaphylactoid rates, and is now used in some UK centers.
Smilkstein Oral Protocol (72-Hour)
Marty Smilkstein's oral protocol was published in the New England Journal of Medicine in 1988. Total dose 1,330 mg/kg over 72 hours:
- Loading: 140 mg/kg orally (dilute the powder in juice or soda to mask the sulfur taste)
- Maintenance: 70 mg/kg orally every 4 hours for 17 additional doses (68 hours total)
If the patient vomits within 1 hour of a dose, that dose must be repeated. Anti-emetics (ondansetron or metoclopramide) are typically given first. Despite the practical difficulty of getting a nauseated overdose patient to keep down 18 doses of foul-smelling NAC over 3 days, the Smilkstein oral protocol is highly effective when completed.
The oral protocol is preferred over IV in three specific scenarios:
- History of anaphylactoid reaction to IV NAC — the oral route does not produce histamine-mediated reactions.
- IV access limited or contaminated — particularly in resource-limited settings or with active IV drug users.
- Patient preference and reliable PO tolerance — some patients tolerate it well, particularly when given as the effervescent preparation with juice.
In modern US practice IV is the default because of (a) faster onset of action, (b) shorter total treatment time, and (c) more reliable dosing in a nauseated patient. Oral is held in reserve for the specific scenarios above.
The 8-Hour Window — Why Timing Is Everything
NAC efficacy is exquisitely time-dependent:
- 0–8 hours post-ingestion: Essentially 100% prevention of hepatotoxicity. Mortality approaches zero. This is the critical window where NAC must be started.
- 8–16 hours: Efficacy declines steeply. Many patients still avoid significant injury but the protective margin shrinks. NAC is still standard of care.
- 16–24 hours: Substantial hepatic injury is increasingly likely; NAC reduces but does not eliminate mortality. Outcomes depend heavily on the size of the ingestion and host susceptibility factors.
- Beyond 24 hours: The role of NAC shifts from prevention of injury to support of recovery. The Lee 2009 Gastroenterology trial showed NAC improves transplant-free survival in established non-acetaminophen acute liver failure as well, so even very late NAC has a role.
The 8-hour boundary is not magic — it reflects the kinetics of NAPQI generation and protein adduct formation. Up to 8 hours, the toxin is still being produced and can still be intercepted. After 8 hours, increasing fractions of NAPQI have already covalently bound to hepatocyte mitochondrial proteins, and these adducts will trigger the apoptotic cascade regardless of whether further toxin is generated.
For patients with unknown ingestion time, the conservative practice is to treat as if the patient is in the 0–8 hour window. The downside of unnecessary NAC is minor (nausea, possible anaphylactoid reaction); the downside of missed treatment is acute liver failure and possible death.
Special Populations and Risk Modifiers
- Chronic alcohol use — lower the treatment threshold. The 150 mcg/mL line on the nomogram may not adequately protect chronic drinkers; some guidelines suggest treating at any detectable acetaminophen level in a chronic alcohol user.
- Isoniazid users — similar logic. CYP2E1 induction lowers the toxic threshold.
- Chronic supratherapeutic dosing — the patient who has been taking 6–8 g/day for several days. No single time-zero, so the nomogram does not apply. Empiric NAC treatment is standard if transaminases are elevated or any acetaminophen is detectable.
- Pregnancy — NAC crosses the placenta and protects the fetal liver. Acetaminophen overdose in pregnancy is treated identically; NAC is FDA pregnancy Category B for this indication.
- Pediatric overdose — same protocols, weight-adjusted dosing. Children actually tend to be somewhat more resistant to acetaminophen toxicity at equivalent mg/kg doses than adults, possibly due to higher constitutive sulfation activity.
- Co-ingestion with opioids — common (Vicodin, Percocet, Norco all contain acetaminophen + opioid). The opioid component delays gastric emptying, extending the absorption window. The 4-hour nomogram level may underestimate peak. Repeat levels and continue NAC if rising.
- Extended-release acetaminophen — the 4-hour nomogram level is unreliable. Treat empirically based on dose ingested.
Adverse Reactions to IV NAC
Anaphylactoid (non-IgE histamine-mediated) reactions occur in approximately 10–20% of patients during the loading dose. The clinical picture: flushing, urticaria, pruritus, occasional bronchospasm, rare hypotension. True anaphylaxis (IgE-mediated) is uncommon. Distinguishing features:
- Anaphylactoid reactions typically occur during the loading infusion (first 15–60 minutes) and resolve when the infusion is paused
- Most patients can resume NAC at a slower rate after a brief pause and antihistamine treatment
- Severity correlates with infusion rate — extending the loading dose from 15 minutes to 60 minutes substantially reduces reaction rates
- Patients with asthma have higher reaction rates and more severe bronchospasm when they occur
Management: pause the infusion, give diphenhydramine 25–50 mg IV, treat bronchospasm with albuterol, then resume at half rate. Reactions rarely require permanent discontinuation; oral NAC is the alternative if IV cannot be tolerated.
Other adverse effects: nausea and vomiting (very common, particularly with oral); sulfurous breath odor; mild headache; transient elevation of INR independent of liver injury (NAC competes with vitamin K for clotting factor synthesis sites — not clinically significant but can confuse interpretation of coagulation studies).
Why NAC Is the Only Proven Antidote
Several other interventions have been tried for acetaminophen overdose. None matches NAC:
- Activated charcoal — effective for adsorption of unabsorbed acetaminophen if given within 1–2 hours of ingestion. Does nothing for absorbed drug. Useful as an adjunct, never as a substitute for NAC. Modern practice often gives a single 1 g/kg dose if the patient presents within 4 hours.
- Methionine — provides sulfur amino acid substrate. Effective when given early but less reliable than NAC because of variable absorption and slower hepatic uptake. Used in some UK protocols historically; largely supplanted by NAC.
- Silymarin (milk thistle) — some animal data for hepatoprotection but no controlled human trials for acetaminophen overdose. Reasonable as a long-term hepatic support during recovery, not as an antidote.
- Glutathione (oral or IV) — oral glutathione has poor bioavailability because it is degraded in the gut. IV glutathione is not standard practice and has never been compared head-to-head with NAC in this indication. NAC is the preferred glutathione delivery vehicle for this purpose.
- Cimetidine — inhibits CYP enzymes. Theoretically attractive but the inhibition is incomplete and onset is slow. Not standard of care.
The mechanism-of-action specificity is what makes NAC irreplaceable here. NAPQI is the toxin; glutathione is the antidote; cysteine is the rate-limiting precursor for glutathione synthesis; and NAC is the only practical way to deliver cysteine to hepatocytes fast enough to keep up with NAPQI generation in overdose.
Cautions
- Do not delay NAC waiting for acetaminophen level if presentation is >8 hours. Start NAC immediately, then de-escalate if the level proves to be below treatment threshold. Time is the enemy.
- Anaphylactoid reactions are not a contraindication to continued NAC treatment. Manage with infusion pause and antihistamine; resume at slower rate.
- This page is for clinical reference only. Acetaminophen overdose is a medical emergency. Anyone who has ingested a toxic dose or believes they may have should call Poison Control (1-800-222-1222 in the US) and go to an emergency department immediately. Do not self-treat with over-the-counter NAC supplements.
- OTC oral NAC supplements (typically 600 mg capsules) are not appropriate substitutes for the 140 mg/kg loading and 70 mg/kg q4h maintenance doses required for overdose. A 70 kg adult would need a 9,800 mg loading dose — sixteen 600 mg capsules — followed by eight more capsules every 4 hours for three days. This must happen in a monitored hospital setting, not at home.
- Chronic acetaminophen risk reduction. Patients with chronic pain who routinely take acetaminophen at therapeutic doses do not need NAC supplementation prophylactically. The hepatic glutathione system handles therapeutic doses without strain. The exception is patients with chronic alcohol use or known CYP2E1 induction, who should discuss long-term acetaminophen use carefully with their physician.
Key Research Papers
- Prescott LF et al. (1979). Intravenous N-acetylcysteine: the treatment of choice for paracetamol poisoning. British Medical Journal. — PubMed
- Smilkstein MJ et al. (1988). Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). New England Journal of Medicine. — PubMed
- Rumack BH, Matthew H (1975). Acetaminophen poisoning and toxicity. Pediatrics. — PubMed
- Mitchell JR et al. (1973). Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. Journal of Pharmacology and Experimental Therapeutics. — PubMed
- Lee WM et al. (2009). Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. — PubMed
- Bateman DN et al. (2014). Reduction of adverse effects from intravenous acetylcysteine treatment for paracetamol poisoning: the SNAP trial. Lancet. — PubMed
- Kerr F et al. (2005). The Australasian Clinical Toxicology Investigators Collaboration randomized trial of different loading infusion rates of N-acetylcysteine. Annals of Emergency Medicine. — PubMed
- Hodgman MJ, Garrard AR (2012). A review of acetaminophen poisoning. Critical Care Clinics. — PubMed
- Heard KJ (2008). Acetylcysteine for acetaminophen poisoning. New England Journal of Medicine review. — PubMed
- Whyte IM et al. (2007). A model for the management of self-poisoning. Medical Journal of Australia. — PubMed
PubMed Topic Searches
- PubMed: NAC acetaminophen overdose
- PubMed: NAPQI CYP2E1 hepatotoxicity
- PubMed: Rumack-Matthew nomogram
- PubMed: acetaminophen chronic alcohol
- PubMed: ALF non-acetaminophen NAC
Connections
- Free Radicals & Your Antioxidant Network — interactive animation
- NAC Overview
- NAC Benefits Hub
- NAC as Glutathione Precursor
- NAC for COPD & Lung
- NAC for Mental Health
- NAC & Liver Health
- NAC & Glutathione
- Glutathione
- Cysteine
- Glycine
- Liver Disease
- Cirrhosis
- NAFLD
- Liver Cleansing
- Detox Protocols
- Oxidative Stress
- All Toxins
- All Antioxidants