Tyrosine for Thyroid Function
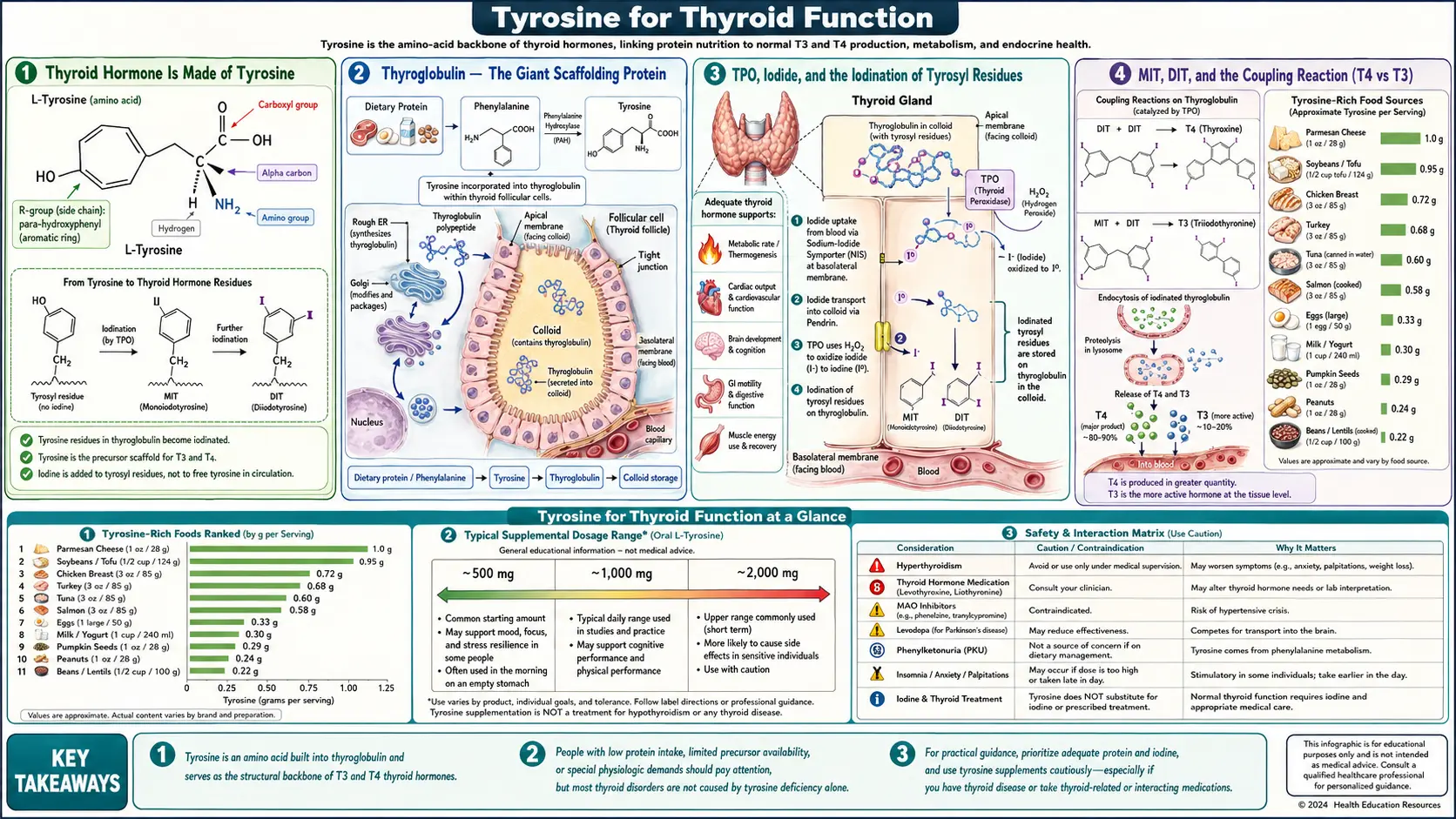
Thyroid hormone is literally made of tyrosine. Thyroxine (T4) is two tyrosine residues joined ether-style with four iodine atoms attached; triiodothyronine (T3) is the same structure with three iodine atoms. Both are assembled in the thyroid gland on a giant scaffolding protein, thyroglobulin, by the enzyme thyroid peroxidase (TPO) acting on iodide and tyrosyl residues. The substrate-availability question for thyroid hormone synthesis therefore has two halves: iodine (the universally limiting micronutrient where deficiency causes goiter and cretinism) and tyrosine (almost never frankly deficient, but a node where supplementation has been explored for subclinical hypothyroidism). This page walks through the biochemistry, the iodine-plus-tyrosine combination logic, and the realistic clinical applications — tyrosine is no substitute for thyroid hormone replacement in primary hypothyroidism, but may have a defensible adjunct role in specific marginal cases.
Interactive Visualization Dopamine, Reward & How Habits Hijack You Dopamine is not the pleasure chemical. Watch the spike migrate from the reward to the cue — then withhold the reward and see it dip below zero. That dip is craving. Launch →
Table of Contents
- Thyroid Hormone Is Made of Tyrosine
- Thyroglobulin — The Giant Scaffolding Protein
- TPO, Iodide, and the Iodination of Tyrosyl Residues
- MIT, DIT, and the Coupling Reaction (T4 vs T3)
- Peripheral Deiodination (T4 → T3 in Tissues)
- Iodine — The Other Half of the Substrate Pair
- Why Tyrosine Deficiency Is Almost Never the Bottleneck
- Subclinical Hypothyroidism — The Plausible Application
- Tyrosine-Plus-Iodine Combination Products
- Hashimoto's Thyroiditis Context
- Cautions — Hyperthyroidism, Graves', Iodine Excess
- Key Research Papers
- Connections
- Featured Videos
Thyroid Hormone Is Made of Tyrosine
This is the most important sentence on the page. The two thyroid hormones — thyroxine (T4) and triiodothyronine (T3) — are not metabolites of tyrosine the way dopamine is, where the original amino acid skeleton is modified and partially preserved. They are made of two whole tyrosine molecules, ether-bonded through their phenolic oxygens, with iodine atoms substituted onto the aromatic rings:
- Thyroxine (T4) — two tyrosine residues, total of four iodine atoms (two on the outer ring, two on the inner ring). Chemical formula C15H11I4NO4. Sometimes written as "3,5,3',5'-tetraiodothyronine" to specify which carbons carry the iodines.
- Triiodothyronine (T3) — the same two-tyrosine structure with three iodines (two on the inner ring, one on the outer ring). Chemical formula C15H12I3NO4. The biologically active form, with 3–5-fold the receptor affinity of T4. The thyroid synthesizes mostly T4; peripheral deiodination converts T4 to T3 in target tissues as needed.
- Reverse T3 (rT3) — same skeleton as T3 but with the iodine in the alternate position on the outer ring (one of the outer iodines, neither of the inner iodines). Biologically inactive at the thyroid hormone receptor; functions essentially as a brake on the active T3 signal. Elevated rT3 is a marker of stress, illness, and impaired peripheral conversion.
The full structural name of T4 reveals the elegance: O-(4-hydroxy-3,5-diiodophenyl)-3,5-diiodo-L-tyrosine. The L-tyrosine backbone is preserved intact, with the second tyrosine grafted onto its phenolic oxygen through an ether bridge. This is the chemistry of every thyroid hormone molecule in human circulation.
The implication: tyrosine is the structural raw material of all thyroid hormone in the body. Without dietary tyrosine (or dietary phenylalanine that the liver converts to tyrosine via phenylalanine hydroxylase), there can be no thyroid hormone synthesis. This is uncontested biochemistry. The clinical question is not whether tyrosine matters for thyroid hormone — it does — but whether tyrosine availability is ever the rate-limiting factor in a healthy diet, and when supplementation might help.
Thyroglobulin — The Giant Scaffolding Protein
Thyroid hormone is not assembled from free tyrosine in solution. It is assembled on tyrosyl residues embedded in a very large secreted protein called thyroglobulin (Tg), which is synthesized by thyroid follicular cells (thyrocytes), secreted into the lumen of thyroid follicles (the spherical cell-bounded compartments that store the hormone), and then iodinated and processed in place.
Key thyroglobulin facts:
- Each thyroglobulin monomer has approximately 2,750 amino acid residues including roughly 110–120 tyrosyl residues
- The functional form is a homodimer of two thyroglobulin chains
- Only a subset (roughly 5–10) of the tyrosyl residues are favorably positioned for iodination and coupling to produce hormone — these are called the "hormonogenic" tyrosines
- Each thyroglobulin dimer can produce approximately 4–5 T4 molecules and small amounts of T3
- Thyroglobulin is stored in the colloid (the central lumen of each thyroid follicle), which is the only significant hormone storage depot in the human endocrine system — the thyroid keeps approximately a 2–3 month supply of hormone in its follicular colloid at any given time
This colloid storage is why TSH-driven hormone release can ramp up rapidly in response to demand — the gland does not need to synthesize new thyroglobulin or iodinate fresh tyrosines; it can simply re-uptake stored thyroglobulin, proteolyze it in lysosomes, and release the embedded T4 and T3 into circulation. The colloid is also why dietary iodine deficiency takes weeks to months to produce frank hypothyroidism — the existing stores must be exhausted first.
The tyrosine relevance: every thyroglobulin molecule the thyroid synthesizes uses approximately 110 tyrosyl residues. The thyroid produces substantial thyroglobulin daily. Severe tyrosine deficiency at the systemic level would impair thyroglobulin synthesis itself, before any question of iodination arises. This effectively never happens in adequately fed adults but is a theoretical consideration in severe protein-energy malnutrition.
TPO, Iodide, and the Iodination of Tyrosyl Residues
The iodination step is catalyzed by thyroid peroxidase (TPO), the enzyme that is targeted by anti-TPO autoantibodies in Hashimoto's thyroiditis and by the anti-thyroid drugs methimazole and propylthiouracil in hyperthyroidism management. The reaction occurs at the apical membrane of the thyrocyte, facing the colloid lumen.
Sequence:
- Iodide uptake — thyrocytes have a basolateral sodium-iodide symporter (NIS) that actively imports iodide from blood against a concentration gradient. The thyroid concentrates iodide to roughly 20–40 times the plasma concentration.
- Iodide transport to lumen — iodide moves through the thyrocyte and exits the apical membrane into the colloid lumen via the pendrin transporter (defects in pendrin cause Pendred syndrome, with hypothyroidism plus sensorineural deafness).
- Iodide oxidation — in the colloid lumen, TPO uses hydrogen peroxide (generated by the dual oxidase enzymes DUOX1 and DUOX2 at the apical membrane) to oxidize iodide to a reactive iodinating species (iodinium-equivalent, often written I+).
- Tyrosyl iodination — the reactive iodine attacks the aromatic ring of tyrosyl residues on thyroglobulin, substituting an iodine for a hydrogen at the 3-position (producing monoiodotyrosine, MIT) and sometimes also at the 5-position (producing diiodotyrosine, DIT). This is also catalyzed by TPO.
- Coupling — TPO then catalyzes the oxidative coupling of two iodinated tyrosyl residues on thyroglobulin to form the thyronine structure: DIT + DIT → T4 (the dominant reaction), MIT + DIT → T3 (smaller fraction), or DIT + MIT in the alternate ether position → rT3.
The product remains covalently attached to thyroglobulin until that thyroglobulin molecule is endocytosed back into the thyrocyte and proteolyzed, releasing free T4, T3, MIT, DIT, and the parent peptide. MIT and DIT are deiodinated by intracellular iodotyrosine deiodinase (IYD), recycling the iodine for reuse.
This is a complex and tightly regulated assembly process. The substrate inputs are iodide and tyrosyl residues on thyroglobulin. The energy input is the hydrogen peroxide oxidant. The catalyst is TPO. Disruption at any node disrupts the system: iodine deficiency, autoimmune TPO destruction (Hashimoto's), Pendred syndrome, congenital TPO deficiency, and iatrogenic methimazole/PTU all impair iodination and coupling.
MIT, DIT, and the Coupling Reaction (T4 vs T3)
The two precursor species — MIT (monoiodotyrosine) and DIT (diiodotyrosine) — combine in different ratios to produce different end products:
- DIT + DIT → T4 (thyroxine, four iodines total) — the dominant product. Approximately 80–90% of thyroid output is T4. The thyroid is essentially a T4 factory under normal conditions; peripheral tissues then deiodinate T4 to T3 on demand.
- MIT + DIT → T3 (triiodothyronine, three iodines total) — the minor product, representing approximately 10–20% of thyroid output. The thyroid does directly secrete some T3, but most circulating T3 comes from peripheral T4 → T3 conversion by deiodinase enzymes.
- DIT + MIT (alternate ether position) → rT3 (reverse triiodothyronine) — trace direct thyroid output, largely a peripheral conversion product from T4. Biologically inactive at the thyroid hormone receptor; functions as a brake on T3 signaling.
The ratio of T4 to T3 produced by the thyroid is influenced by iodine availability: under iodine sufficiency, DIT residues are abundant and DIT + DIT → T4 dominates; under mild iodine restriction, MIT residues are relatively more abundant and the MIT + DIT → T3 fraction increases. This is an adaptive mechanism that prioritizes the biologically active T3 over the less-active T4 when iodine is scarce.
The tyrosyl precursors here are not "free tyrosine in the cytosol" — they are tyrosyl residues at specific positions on the thyroglobulin scaffold. The relevant substrate availability question is "how much thyroglobulin can the thyroid synthesize and how many of its tyrosyl residues can be iodinated", not "what is the systemic plasma tyrosine concentration". This is why simple tyrosine supplementation does not straightforwardly increase thyroid hormone output in normal subjects — the substrate is not the systemic tyrosine pool but the localized thyroglobulin-tyrosyl pool.
Peripheral Deiodination (T4 → T3 in Tissues)
Most of circulating T4 is converted to T3 (or to inactive rT3) in peripheral tissues by a family of three selenium-containing deiodinase enzymes:
- Type 1 deiodinase (D1) — expressed in liver, kidney, thyroid. Converts T4 to T3 (outer-ring deiodination, "activation") and converts rT3 to T2 (clearance). Major contributor to circulating T3 pool. Selenium-dependent.
- Type 2 deiodinase (D2) — expressed in brain, pituitary, brown adipose tissue, skeletal muscle, thyroid. Converts T4 to T3 locally inside target cells, producing intracellular T3 for nuclear receptor binding. Selenium-dependent. Critical for brain T3 supply.
- Type 3 deiodinase (D3) — expressed in placenta, fetal tissues, brain, skin. Converts T4 to rT3 and T3 to T2 (both inner-ring deiodinations, "inactivation"). Acts as a brake on T3 signaling. Selenium-dependent.
The clinical implication: peripheral T4 → T3 conversion depends critically on selenium adequacy. Selenium deficiency is associated with elevated TSH, elevated rT3, and clinical hypothyroid features even when thyroid output is normal — the gland is making hormone, but the body can't convert T4 to active T3. This is why our Selenium page emphasizes the thyroid connection and why selenium repletion is a routine consideration in workup of suboptimal T3 with normal T4.
The deiodination pathway is also stress-sensitive. Severe illness, prolonged fasting, anorexia nervosa, and chronic inflammation shift the balance toward rT3 production (via increased D3 activity and decreased D1 activity) — the "low T3 syndrome" or "nonthyroidal illness syndrome" common in hospitalized critically ill patients. This is generally an adaptive response and does not require T3 supplementation; correcting the underlying illness restores normal deiodination.
Tyrosine has no direct role at the peripheral deiodination step — this is iodine and selenium biochemistry on a pre-existing T4 molecule. Tyrosine supplementation will not improve T4 → T3 conversion in a selenium-deficient patient.
Iodine — The Other Half of the Substrate Pair
Iodine is the universally limiting micronutrient for thyroid hormone synthesis worldwide, with billions of people in iodine-deficient regions historically affected before the universal salt iodization programs of the 20th century. The thyroid is the principal sink for dietary iodine in humans, concentrating approximately 70–80% of body iodine in a gland weighing 20–30 grams.
Iodine status determinants:
- Adequate dietary intake — the WHO RDA is 150 mcg/day for non-pregnant adults, 220 mcg/day in pregnancy, 290 mcg/day during lactation
- Iodized salt programs — the largest single nutritional intervention in human history, credited with eliminating endemic goiter from most of the developed world. Iodized salt provides approximately 45 mcg iodine per gram of salt.
- Seafood, dairy, and eggs — the major non-salt dietary iodine sources. Seafood is the densest natural source (haddock, cod, tuna, kelp). Dairy contributes substantially in some regions through iodophor sanitizers used on milking equipment.
- Goitrogens — thiocyanate (from cruciferous vegetables and from cyanogenic glycosides in cassava), perchlorate (industrial contaminant), and nitrate (from fertilizer runoff in groundwater) all compete with iodide for NIS uptake. Generally clinically insignificant unless combined with marginal iodine intake.
- Excessive iodine — can paradoxically suppress thyroid function via the Wolff-Chaikoff effect (acute) or precipitate Jod-Basedow hyperthyroidism in autonomous nodular thyroid disease. The upper-intake limit is 1,100 mcg/day for adults.
If iodine intake is inadequate, no amount of tyrosine supplementation will support adequate thyroid hormone production — iodine is the missing ingredient. Conversely, in iodine-replete subjects, tyrosine status is essentially always adequate from dietary protein, and supplementation produces no additional thyroid hormone output. This is the fundamental constraint that limits tyrosine supplementation as a thyroid intervention: in iodine-deficient populations, iodine itself is the answer; in iodine-replete populations, neither iodine nor tyrosine is limiting.
See our Iodine page for the full nutritional biochemistry of iodine, the history of iodized salt programs, and the dietary sources.
Why Tyrosine Deficiency Is Almost Never the Bottleneck
In a healthy adult eating a normal mixed diet:
- Dietary tyrosine intake from protein-containing foods is typically 1–5 g/day
- The liver additionally synthesizes tyrosine from phenylalanine via PAH at a substantial rate
- Plasma tyrosine concentration is maintained in a narrow range (40–80 µmol/L)
- The total body tyrosine pool turnover is on the order of grams per day
- Thyroglobulin synthesis consumes a comparatively trivial amount of tyrosine per day — on the order of milligrams
The asymmetry is large. The thyroid's tyrosine requirement is a tiny rounding error compared to the systemic tyrosine flux from dietary intake and from phenylalanine conversion. Frank tyrosine deficiency limiting thyroid hormone synthesis essentially does not occur in adequately fed adults.
The theoretical contexts where it could occur:
- Severe protein-energy malnutrition (marasmus, kwashiorkor) — affects every protein-dependent process including thyroglobulin synthesis
- Untreated phenylketonuria (PKU) with elevated phenylalanine — the elevated phenylalanine competes for transporters and may reduce tyrosine availability; treated PKU diets include tyrosine supplementation
- Very-low-protein diets (under 0.5 g protein/kg/day) sustained for months — theoretically marginal but rarely clinically symptomatic for thyroid specifically
- Severe liver disease impairing PAH activity — rare
None of these contexts is improved by adding 1–2 g of supplemental tyrosine to an otherwise inadequate diet; the underlying nutritional problem must be addressed comprehensively.
So why does tyrosine appear in so many "thyroid support" supplement products? Largely because the biochemistry is intuitive (tyrosine is the building block!) and the supplement industry markets to that intuition. The clinical evidence for tyrosine adding value to iodine-and-selenium-and-zinc-replete adults with primary hypothyroidism is thin to none.
Subclinical Hypothyroidism — The Plausible Application
Subclinical hypothyroidism is the laboratory state of elevated TSH (typically 4–10 mIU/L) with free T4 still in the normal range. It affects approximately 5–10% of adults, with higher prevalence in women, older adults, and patients with Hashimoto's thyroiditis. Whether and when to treat subclinical hypothyroidism with levothyroxine remains a clinical judgment call, with most guidelines recommending treatment for TSH above 10 mIU/L and individualized decisions for TSH 4–10 mIU/L.
In this gray zone, an empirical trial of tyrosine plus iodine plus selenium and other micronutrients is sometimes considered by integrative practitioners, with the goal of optimizing substrate and cofactor availability before committing to lifelong levothyroxine. The reasoning:
- If subclinical hypothyroidism reflects a marginal substrate problem (mild iodine deficiency in a non-iodized-salt area, marginal selenium, marginal tyrosine), addressing all of those simultaneously may normalize TSH
- If subclinical hypothyroidism reflects early Hashimoto's (anti-TPO positive, gradual gland destruction), micronutrient support will not reverse the autoimmune process but may delay progression and is unlikely to do harm
- If subclinical hypothyroidism reflects nonthyroidal illness (chronic inflammation, severe stress, recent illness), addressing the underlying condition is more important than micronutrient support
A defensible micronutrient stack for subclinical hypothyroidism trial (not a treatment recommendation; coordinate with your clinician):
- L-tyrosine 500–1000 mg in the morning, on empty stomach
- Iodine (kelp-derived or potassium iodide) 150–300 mcg/day if intake assessment suggests inadequacy
- Selenium 100–200 mcg/day (selenomethionine preferred)
- Zinc 15–30 mg/day if status low (zinc supports TSH receptor function and deiodinase activity)
- Iron repletion if ferritin below 50 ng/mL (iron deficiency suppresses TPO activity)
- Vitamin D repletion to 30–50 ng/mL
- Reassess TSH and free T4 at 8–12 weeks; if no improvement, escalate to levothyroxine consideration
This is a reasonable trial in a patient who prefers to attempt micronutrient optimization first. It is not a substitute for thyroid hormone replacement in established overt hypothyroidism (TSH >10 with low free T4 and clinical symptoms).
See our Hashimoto's Thyroiditis page for the full autoimmune-thyroid clinical context and our Reverse T3 and Low T3 Syndrome sub-article for the conversion-pathway considerations.
Tyrosine-Plus-Iodine Combination Products
Several proprietary supplement formulations combine L-tyrosine with iodine (typically as kelp extract or potassium iodide) marketed for thyroid support. The biochemical logic is straightforward — pair the two substrate inputs to thyroid hormone synthesis. The clinical evidence for these combinations producing measurable hormone elevation in iodine-and-tyrosine-replete adults is thin, but the combination is defensible in marginal-iodine populations and in subclinical hypothyroidism trials as discussed above.
Typical dosing in such products:
- L-tyrosine 500–1000 mg
- Iodine 150–300 mcg (kelp extract or potassium iodide)
- Sometimes also selenium, zinc, copper, manganese, vitamin A as additional thyroid-relevant micronutrients
Practical considerations:
- Take in the morning on empty stomach; avoid concurrent dosing with calcium, iron, or thyroid medication (which compete for absorption)
- If on levothyroxine, separate the supplement from the levothyroxine by 4 hours to avoid binding interactions
- Monitor TSH at 8–12 weeks of consistent dosing to assess response
- Discontinue if TSH suppresses below 0.5 mIU/L (suggests overshoot)
- Discontinue if hyperthyroid symptoms emerge (palpitations, tremor, heat intolerance, weight loss)
Hashimoto's Thyroiditis Context
Hashimoto's thyroiditis is the most common cause of hypothyroidism in iodine-replete populations. The autoimmune destruction targets thyroid peroxidase (TPO) and thyroglobulin, gradually reducing the gland's capacity to synthesize hormone. Once the autoimmune process is established, tyrosine and iodine substrate availability are not the rate-limiting factors — functional thyroid tissue is.
In Hashimoto's, supplemental tyrosine will not slow the autoimmune process and will not restore lost thyroid function. The role of supplementation is restricted to:
- Subclinical phase (anti-TPO positive, TSH still normal) — substrate and cofactor support is benign and may modestly delay progression
- Mild early hypothyroidism (TSH 4–10) — trial as discussed in subclinical hypothyroidism section
- Established hypothyroidism on levothyroxine — tyrosine may have a role in supporting the noradrenergic stress response that is often abnormal in hypothyroid patients (fatigue, low mood, brain fog), independent of the thyroid hormone replacement
The autoimmune trigger and management of Hashimoto's involves selenium (anti-TPO antibody titer reduction in some trials), vitamin D, gluten and food-trigger investigation, gut microbiome assessment, and reducing oxidative stress — these are addressed comprehensively on our Hashimoto's Thyroiditis page. Tyrosine is at most a small piece of the puzzle in Hashimoto's.
Cautions — Hyperthyroidism, Graves', Iodine Excess
- Active hyperthyroidism or Graves' disease — do not use tyrosine. The gland is already overproducing hormone; providing additional substrate is contraindicated. Wait until the hyperthyroid state is medically controlled (with methimazole, PTU, radioactive iodine, or thyroidectomy) before considering any thyroid-targeted supplementation.
- Toxic multinodular goiter — same caution as Graves'. Autonomous nodules can be triggered into excessive hormone production by iodine loading (Jod-Basedow phenomenon). Tyrosine plus iodine combinations are particularly risky.
- Excessive iodine intake — consuming more than 1,100 mcg/day of iodine (the upper-intake limit) can paradoxically suppress thyroid function via the Wolff-Chaikoff effect, especially in those with autoimmune thyroid disease. Some thyroid-support supplements stack kelp extract on top of dietary iodized salt and seafood intake to a total that exceeds the safe upper limit.
- Levothyroxine therapy — do not co-dose tyrosine and iodine supplements with the morning levothyroxine. Separate by at least 4 hours to avoid binding interactions and to permit accurate TSH titration. Inform the prescribing clinician of any supplement use to allow appropriate TSH monitoring.
- Pregnancy — pregnancy increases thyroid hormone requirement by approximately 50%. Iodine requirements increase to 220 mcg/day. Tyrosine intake from a normal protein-adequate diet is sufficient. Self-supplementation with high-dose tyrosine or with iodine-tyrosine combinations during pregnancy should be coordinated with the prenatal care provider; both excess and deficiency of iodine in pregnancy have adverse fetal effects (cretinism from deficiency, fetal hypothyroidism from excess).
- Postpartum thyroiditis — transient hyperthyroid phase (1–6 months postpartum) followed by transient or permanent hypothyroid phase. Avoid thyroid-stimulating supplements during the hyperthyroid phase; tyrosine and iodine may be relevant after thyroid function normalizes or in the hypothyroid phase, coordinated with the clinician.
- Drug interactions — methimazole, propylthiouracil, lithium, and amiodarone all interact with thyroid hormone synthesis. Do not add tyrosine or iodine supplementation to these medications without clinical guidance.
- Concomitant levothyroxine titration — starting tyrosine + iodine in a patient already on stable levothyroxine can cause TSH to drift, requiring dose adjustment. Inform the prescriber and arrange TSH monitoring at 6–8 weeks.
Key Research Papers
- Carvalho DP, Dupuy C (2017). Thyroid hormone biosynthesis and release. Molecular and Cellular Endocrinology. — PubMed
- Ruf J, Carayon P (2006). Structural and functional aspects of thyroid peroxidase. Archives of Biochemistry and Biophysics. — PubMed
- Di Jeso B, Arvan P (2016). Thyroglobulin from molecular and cellular biology to clinical endocrinology. Endocrine Reviews. — PubMed
- Bianco AC et al. (2002). Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocrine Reviews. — PubMed
- Zimmermann MB (2009). Iodine deficiency. Endocrine Reviews. — PubMed
- Pearce EN et al. (2013). Global iodine nutrition: where do we stand in 2013? Thyroid. — PubMed
- Garber JR et al. (2012). Clinical practice guidelines for hypothyroidism in adults. Endocrine Practice (AACE/ATA). — PubMed
- Surks MI, Sievert R (1995). Drugs and thyroid function. New England Journal of Medicine. — PubMed
- Drutel A et al. (2013). Selenium and the thyroid gland: more good news for clinicians. Clinical Endocrinology. — PubMed
- Toulis KA et al. (2010). Selenium supplementation in the treatment of Hashimoto's thyroiditis: a systematic review and a meta-analysis. Thyroid. — PubMed
- Maxon HR, Saenger EL (1977). Effects of acute iodine administration on thyroid function: implications for radiation dose to the thyroid. Journal of Nuclear Medicine. — PubMed
- Pearce EN, Braverman LE (2009). Environmental pollutants and the thyroid. Best Practice & Research Clinical Endocrinology & Metabolism. — PubMed
- Stathatos N (2012). Thyroid physiology. Medical Clinics of North America. — PubMed
PubMed Topic Searches
- PubMed: Tyrosine and thyroid hormone synthesis
- PubMed: TPO iodination reaction
- PubMed: Thyroglobulin iodination
- PubMed: Subclinical hypothyroidism management
- PubMed: Iodine and tyrosine for thyroid
Connections
- Dopamine, Reward & How Habits Hijack You — interactive animation
- Tyrosine Overview
- Tyrosine Benefits Hub
- Tyrosine for Stress & Cognitive Performance
- Tyrosine for Dopamine & Mood
- Tyrosine for Cold Tolerance
- Phenylalanine (Precursor)
- Iodine (Substrate Pair)
- Selenium (Deiodinase Cofactor)
- Zinc
- Iron (TPO Cofactor)
- Hashimoto's Thyroiditis
- Reverse T3 and Low T3 Syndrome
- Thyroid Panel
- Fatigue
- Vitamin A (Thyroid Receptor)
- All Amino Acids