Methionine for Detoxification
The body's capacity to clear heavy metals, hormones, drug metabolites, and environmental xenobiotics depends critically on sulfur supply, and the dominant entry point for sulfur into the body is dietary methionine. Through the transsulfuration pathway, methionine becomes cysteine, which then becomes glutathione (the master substrate for Phase II conjugation), metallothionein (the principal heavy metal-binding protein), and a substantial fraction of the sulfated glycosaminoglycans, sulfated steroid metabolites, and inorganic sulfate that the kidneys use to clear water-soluble toxins. SAMe itself (the methionine-derived methyl donor) is the cofactor for the arsenic methyltransferase that detoxifies inorganic arsenic, the catechol-O-methyltransferase that clears catecholamines and catecholestrogens, and the histamine N-methyltransferase that inactivates intracellular histamine. This deep-dive walks through the methionine-to-detoxification pipeline in detail — the transsulfuration enzymes, the glutathione conjugation pathway, the metallothionein and inorganic-sulfate routes, and the specific clinical pictures (heavy metal exposure, estrogen-dominance symptoms, histamine intolerance, multiple chemical sensitivity) where methionine-pathway support is part of an integrative-medicine approach.
Table of Contents
- Phase I and Phase II Liver Detoxification
- The Methionine-Cysteine-Glutathione Pipeline
- Glutathione Conjugation (GST Enzymes)
- Sulfation (PST/SULT) and Inorganic Sulfate
- Metallothionein and Heavy Metal Binding
- Mercury, Lead, Cadmium — Specific Mechanisms
- Arsenic and SAMe-Dependent Methylation
- Estrogen Metabolism via COMT
- Histamine Clearance and HNMT
- Clinical Detoxification Applications
- Cautions in Detoxification Protocols
- Key Research Papers
- Connections
- Featured Videos
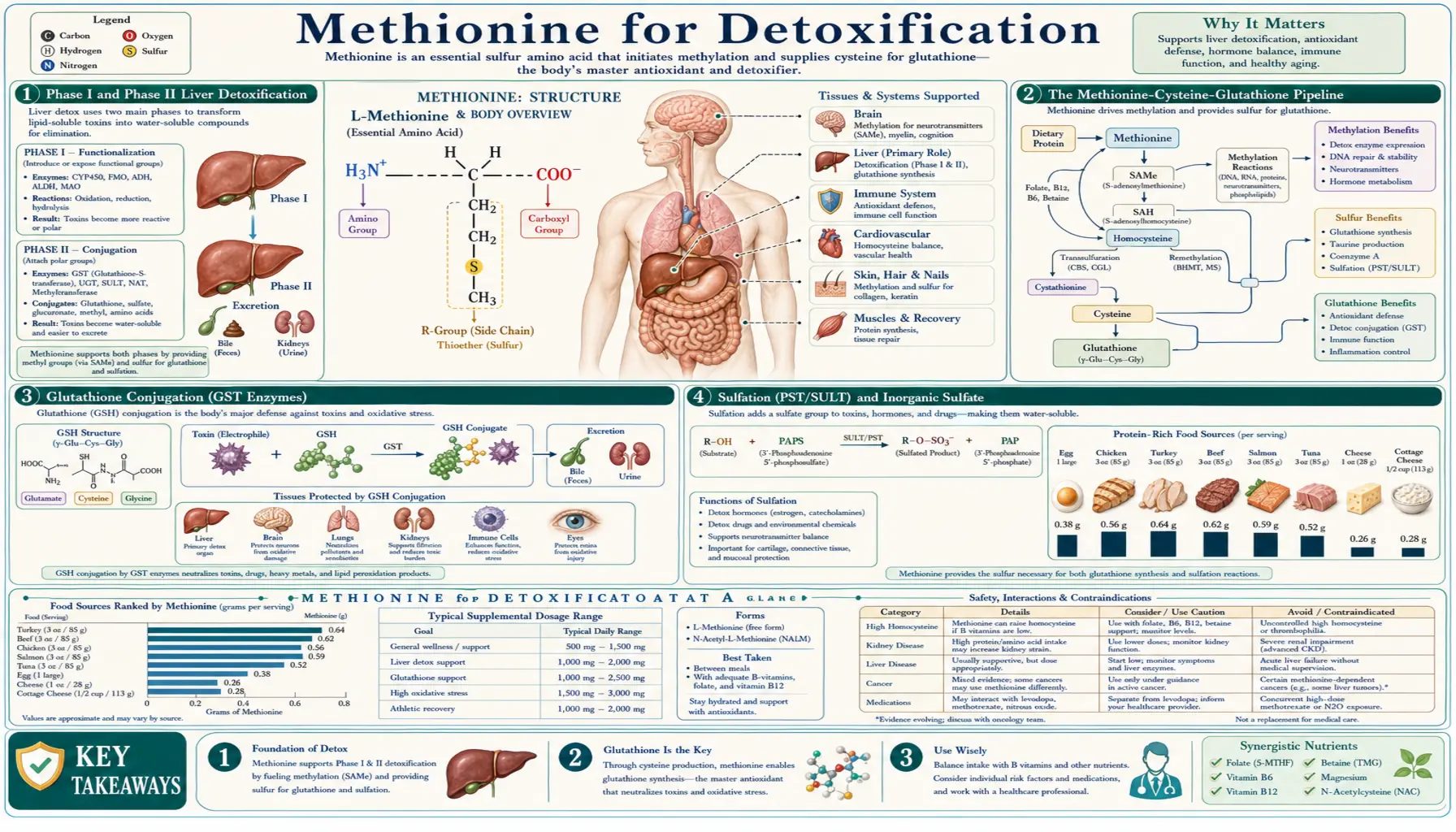
Phase I and Phase II Liver Detoxification
The conventional pharmacology-textbook model divides hepatic xenobiotic clearance into two sequential phases:
- Phase I — functionalization: cytochrome P450 enzymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and dozens of others) introduce or expose polar functional groups on the parent compound through oxidation, reduction, or hydrolysis. The products are often more reactive than the parents because Phase I commonly generates electrophilic intermediates that can damage cellular macromolecules if not immediately conjugated.
- Phase II — conjugation: large hydrophilic groups are appended to the Phase I product (or to the parent compound directly, for substances that bypass Phase I), increasing water solubility and enabling biliary or urinary excretion. The principal Phase II reactions are glutathione conjugation, glucuronidation, sulfation, acetylation, methylation, and glycine conjugation.
The methionine pathway feeds at least four of these Phase II reactions:
- Glutathione conjugation — glutathione synthesis depends on cysteine, which is derived from methionine via transsulfuration
- Sulfation — the PAPS sulfate donor depends on inorganic sulfate ultimately derived from cysteine catabolism (which is downstream of methionine)
- Methylation — SAMe (methionine-derived) is the methyl donor for COMT, HNMT, AS3MT, TPMT, and other Phase II methyltransferases
- Glycine conjugation — while not directly methionine-dependent, glycine availability is linked to the broader sulfur amino acid pool because glycine and glutamate are the other two amino acids in glutathione
The clinical implication is that detoxification capacity is not a single function but a set of parallel pathways, each with its own substrate, cofactor, and rate-limiting step. Methionine deficiency selectively impairs the sulfur-dependent and methylation-dependent routes (glutathione, sulfation, methylation), while leaving glucuronidation and acetylation relatively intact. The result is that substances normally cleared via the sulfur and methylation routes accumulate — mercury, arsenic, certain pesticides, catecholestrogens, histamine, and a substantial subset of pharmaceuticals.
For the conventional pharmacology framing of Phase I and Phase II, see our Detoxification page.
The Methionine-Cysteine-Glutathione Pipeline
The conversion of methionine to glutathione proceeds through a well-characterized enzymatic sequence:
- Methionine + ATP to SAMe (MAT enzyme) — the methionine activation step
- SAMe to SAH via methyltransferase action (any of more than 200 SAMe-using enzymes)
- SAH to homocysteine + adenosine (SAHH/AHCY)
- Homocysteine + serine to cystathionine (cystathionine beta-synthase, CBS, requires vitamin B6 as PLP). This is the committing step of the transsulfuration pathway — once homocysteine is converted to cystathionine, the sulfur is irreversibly committed to cysteine and cannot return to methionine.
- Cystathionine to cysteine + alpha-ketobutyrate (cystathionine gamma-lyase, CGL/CTH, also requires B6 as PLP)
- Cysteine + glutamate to gamma-glutamylcysteine (glutamate-cysteine ligase, GCL, the rate-limiting step of glutathione synthesis)
- Gamma-glutamylcysteine + glycine to glutathione (GSH) (glutathione synthetase)
Three points warrant emphasis:
- The transsulfuration step is regulated by SAMe — CBS is allosterically activated by SAMe and inhibited by accumulated SAH. When SAMe is abundant (signaling adequate methionine supply for methylation needs), CBS is active and shunts excess homocysteine toward cysteine. When SAMe is depleted, CBS is inactive and homocysteine is preferentially recycled back to methionine through the remethylation pathway. This SAMe-mediated regulation ensures that methylation demand is met first, with excess methionine flowing to cysteine and glutathione.
- Cysteine is the rate-limiting substrate for glutathione — while all three amino acids (cysteine, glutamate, glycine) are required for glutathione synthesis, cysteine is by far the least abundant in the intracellular pool, and its availability determines glutathione synthesis rate. This is why supplemental N-acetylcysteine (NAC) is the standard way to acutely boost glutathione — NAC bypasses the entire methionine cycle and delivers cysteine directly.
- The glutathione pool is dynamic and rapidly turning over — hepatic glutathione has a half-life of approximately 90 minutes. Continuous synthesis from cysteine substrate is required to maintain the pool, which is why short-term cysteine depletion (severe acetaminophen overdose, prolonged fasting, severe oxidative stress) can rapidly exhaust the glutathione pool even with normal upstream methionine intake.
The implication for detoxification practice is that boosting glutathione during a high-demand period (chemical exposure, infectious illness with high oxidative stress, intentional detoxification protocol) requires both adequate methionine for the upstream supply and adequate cysteine substrate (typically NAC supplementation) for the immediate synthesis demand. Glycine is generally available in adequate amounts but bone broth or supplemental glycine 3-5 g/day is a reasonable adjunct in clinical detoxification protocols.
Glutathione Conjugation (GST Enzymes)
Glutathione S-transferases (GSTs) are a large family of Phase II enzymes that catalyze the conjugation of reduced glutathione to electrophilic substrates. The human genome encodes approximately 17 cytosolic GST isoforms across seven gene families (alpha, mu, pi, theta, zeta, omega, sigma), plus several membrane-bound microsomal GST isoforms. Different isoforms have different tissue distributions and substrate specificities:
- GSTA (alpha class) — highly expressed in liver and kidney. Handles steroid hormones, cumene hydroperoxide, and many environmental electrophiles.
- GSTM (mu class) — includes GSTM1, which has a null polymorphism (GSTM1*0) in roughly 50% of the population (no functional protein produced). GSTM1-null individuals have measurably reduced clearance of certain environmental electrophiles and modestly elevated cancer risk for several exposures.
- GSTP (pi class) — highly expressed in lung and placenta. Detoxifies polycyclic aromatic hydrocarbons (PAHs from combustion exposure) and many chemotherapy agents.
- GSTT (theta class) — includes GSTT1, which similarly has a common null polymorphism (GSTT1*0) in roughly 20% of the population.
The functional output of GST-mediated conjugation is a glutathione-substrate conjugate that is then processed through several intermediate steps (loss of the gamma-glutamyl residue, loss of the glycine, N-acetylation of the cysteine residue) to yield a mercapturic acid. Mercapturic acids are highly water-soluble and are excreted in urine. Quantification of urinary mercapturic acids is the most sensitive biomarker of in-vivo Phase II glutathione conjugation activity.
The substrates handled by GST-mediated conjugation include:
- Reactive oxygen and electrophilic stress products — 4-hydroxynonenal (lipid peroxidation product), reactive aldehydes
- Polycyclic aromatic hydrocarbons — from cigarette smoke, vehicle exhaust, charred meat (benzo[a]pyrene and many congeners after Phase I activation)
- Heterocyclic amines — formed from amino acids and creatine in high-temperature cooking of meat
- Acetaminophen-derived NAPQI — the supratherapeutic-dose metabolite of acetaminophen
- Many pharmaceuticals and their Phase I metabolites — including ethacrynic acid, busulfan, chlorambucil, melphalan, cisplatin (partial)
- Estrogen quinones — the 4-hydroxyestrogen quinone is a reactive electrophile implicated in estrogen-dependent carcinogenesis; GST conjugation is part of its clearance route
- Heavy metal complexes — particularly inorganic mercury and methylmercury, which form glutathione adducts that are exported via canalicular MRP2 to bile
Adequate methionine intake feeds the upstream cysteine supply that maintains the glutathione pool that the GST enzymes consume during conjugation. Severe methionine restriction, with concurrent inadequate dietary cysteine and NAC, can produce a state of chronic glutathione depletion that impairs Phase II conjugation broadly. This is one mechanism behind the observation that severely undernourished populations have elevated susceptibility to environmental chemical injury.
Sulfation (PST/SULT) and Inorganic Sulfate
Sulfation is a major Phase II conjugation pathway that attaches inorganic sulfate (SO3) to hydroxyl, amine, or other nucleophilic groups on the substrate. The active sulfate donor is 3'-phosphoadenosine-5'-phosphosulfate (PAPS), and the enzymes are the sulfotransferase (SULT, also called PST for phenol sulfotransferase) family, which includes approximately 13 isoforms in humans.
Sulfation is the dominant Phase II route for several physiologically important classes of substrate:
- Sulfated steroid hormones — dehydroepiandrosterone sulfate (DHEA-S), estrone sulfate, pregnenolone sulfate. Sulfation makes the steroid water-soluble for storage and excretion, and is a reversible reaction (sulfatases regenerate the active steroid in target tissues).
- Sulfated bile acids — cholate-sulfate, chenodeoxycholate-sulfate are produced in liver disease as protective alternatives to unconjugated bile acids
- Sulfated catecholamines and their metabolites — dopamine-sulfate, norepinephrine-sulfate, and the COMT product 3-methoxytyramine-sulfate
- Phenolic xenobiotics — acetaminophen at therapeutic doses (10-15% goes through sulfation), bisphenol A (BPA), many food phytochemicals, neurotransmitter precursors and analogs
- Sulfated glycosaminoglycans — chondroitin sulfate, heparan sulfate, dermatan sulfate, keratan sulfate, and heparin all incorporate sulfate from the same PAPS pool used for xenobiotic sulfation
The inorganic sulfate supply for PAPS synthesis comes from two sources:
- Direct dietary sulfate — small contribution from dietary sulfate in vegetables, some mineral waters, and certain food additives
- Cysteine catabolism via the cysteine dioxygenase (CDO) pathway — the major source. Cysteine is oxidized to cysteine sulfinic acid, then to taurine (with the sulfur released as sulfite, then sulfate), or alternatively to pyruvate (with direct release of inorganic sulfate). Both pathways contribute the inorganic sulfate that feeds PAPS synthesis.
Because the cysteine catabolism pathway is the major source of inorganic sulfate, and cysteine is itself derived from methionine via transsulfuration, the methionine pathway feeds the sulfation pathway as well as the glutathione pathway. Severe methionine restriction can reduce PAPS availability and impair sulfation-dependent Phase II conjugation. This has been demonstrated in animal models and in human cases of severe protein-calorie undernutrition.
Clinically, the patients most at risk of impaired sulfation are those with restricted-protein diets (chronic kidney disease, advanced liver disease), molybdenum deficiency (molybdenum is the essential cofactor for sulfite oxidase, the final step in sulfate production from cysteine catabolism), and certain genetic disorders of sulfite oxidase or molybdenum cofactor synthesis. Many integrative-medicine practitioners use supplemental epsom salt baths (magnesium sulfate, with a topical contribution to sulfate status) and magnesium sulfate oral or intravenous solutions in patients with suspected sulfation insufficiency.
For the molybdenum-and-detoxification connection, see our Molybdenum and Detoxification page.
Metallothionein and Heavy Metal Binding
Metallothionein (MT) is a small cysteine-rich protein (approximately 61 amino acids, of which 20 are cysteine) that binds heavy metal ions through coordination to its cysteine thiol groups. Each MT molecule can bind up to seven zinc or copper ions through clusters of cysteine residues forming tetrahedral metal-thiolate cages. MT is the body's principal acute heavy-metal-binding protein and serves as both a zinc storage pool and as the immediate sequestration mechanism for toxic divalent metals (cadmium, mercury, lead, less efficiently arsenic).
Four mammalian MT isoforms exist (MT1, MT2, MT3, MT4) with overlapping but distinct tissue distributions. Hepatic and renal MT (predominantly MT1 and MT2) is induced by exposure to heavy metals, oxidative stress, glucocorticoids, cytokines, and zinc itself. The induction process is rapid — new MT protein appears within hours of metal exposure — and is the body's first-line defense against acute heavy metal toxicity.
The dependence on cysteine is absolute. Without adequate cysteine supply, the body cannot synthesize new MT to bind incoming metal ions, and the burden falls on the limited preexisting MT pool. Methionine deficiency reduces cysteine supply via the transsulfuration pathway, secondarily reducing MT synthesis capacity. This has been demonstrated in animal models of metal toxicity, where methionine-restricted animals show accelerated tissue accumulation and toxicity from cadmium or mercury exposure compared to methionine-replete animals.
Beyond MT, the body's heavy-metal handling depends on at least four other sulfur-containing systems:
- Glutathione — binds methylmercury and inorganic mercury through its cysteine thiol; conjugates are exported via MRP2 to bile and reabsorbed in the entero-hepatic circulation (which is why mercury depuration is slow and why oral chelators that disrupt enterohepatic recirculation can accelerate clearance)
- Glutathione conjugates of organomercurials — methylmercury-glutathione is the principal blood-transport form of methylmercury and the substrate for active uptake into the brain via the LAT1 amino acid transporter (recognized as a methionine analog)
- Selenoproteins — selenium and mercury have high affinity for each other; the selenide ion (Se2-) reacts with mercury to form insoluble mercury selenide that is biologically inert. This is the basis for the apparent protective effect of selenium against mercury toxicity, particularly in high-fish-consumption populations where selenium and methylmercury intake are both high.
- Albumin sulfhydryl groups — serum albumin's single free cysteine (Cys34) is a significant pool of plasma thiol that binds and transports heavy metals.
For clinical heavy metal exposure management, see the section on Mercury, Lead, Cadmium below and our Heavy Metals page.
Mercury, Lead, Cadmium — Specific Mechanisms
The three most commonly encountered toxic heavy metals in modern clinical practice have distinct toxicokinetic patterns but converge on the same sulfur-handling pathway for clearance:
- Mercury — exists in elemental (Hg0, dental amalgam vapor exposure), inorganic divalent (Hg2+, occupational and food contamination), and organic (methylmercury, ethylmercury) forms. Methylmercury from fish (predator species: tuna, swordfish, shark, king mackerel, tilefish) is the dominant exposure for most adults. Methylmercury is absorbed nearly completely from the GI tract, distributes widely including across the blood-brain barrier (LAT1 amino acid transporter recognizes methylmercury-cysteine as a methionine analog and transports it actively into the brain), and is cleared slowly with a biological half-life of 50-70 days. The dominant clearance route is glutathione conjugation in the liver, biliary excretion, and partial deconjugation in the gut with reabsorption (entero-hepatic recirculation, which prolongs the half-life). Adequate methionine for glutathione supply is necessary but not sufficient for accelerated clearance; oral chelators (DMSA, DMPS) and reduction of ongoing exposure (fish dietary modification) are the primary interventions.
- Lead — primary modern exposures are old paint (pre-1978 housing), contaminated drinking water, certain imported pottery and cosmetics, and occupational exposures (battery manufacturing, demolition, shooting ranges). Lead is absorbed less completely than mercury (10-15% in adults, 40-50% in children), distributes initially to soft tissue and then to bone (where it has a 20-30 year half-life), and competes with calcium and zinc at multiple enzyme sites. Acute lead poisoning is treated with EDTA or DMSA chelation. Chronic low-level exposure is managed by source reduction, calcium and iron adequacy (which reduce lead absorption), and supportive nutrition including methionine and antioxidant pathway support. Glutathione conjugation is not the primary clearance route for lead (most lead is renally excreted unchanged), but glutathione protects against the secondary oxidative stress that lead induces.
- Cadmium — principal exposures are cigarette smoking (each cigarette delivers approximately 1-2 micrograms of inhaled cadmium with high lung absorption), and dietary sources (leafy greens grown in cadmium-contaminated soil, organ meats, certain shellfish). Cadmium has an exceptionally long biological half-life (10-30 years in the kidney). MT binding sequesters cadmium in the kidney where it concentrates over a lifetime; once renal MT is saturated, cadmium toxicity manifests as proximal tubular dysfunction and progressive kidney disease. Cadmium has no efficient chelation therapy; clinical management is exposure avoidance and supportive nutrition.
The clinical pattern that brings patients to integrative-medicine attention for suspected heavy metal toxicity includes chronic fatigue, cognitive dysfunction, immune dysregulation, peripheral neuropathy, mood disturbance, autoimmunity, and skin findings (in mercury particularly, the older mad-hatter pattern with tremor, irritability, and gingivitis). Definitive evaluation requires whole-blood mercury, urine mercury (with or without DMSA challenge for tissue-burden assessment, with caveat that the challenge test is interpretively complex), and consultation with a clinical toxicologist or environmental medicine specialist for significant exposures. Methionine-pathway support (adequate dietary methionine, supplemental NAC, selenium adequacy, glutathione-supportive cofactors) is a sensible part of integrative management but does not substitute for source identification and specialist chelation in significant exposures.
Arsenic and SAMe-Dependent Methylation
Inorganic arsenic (drinking water contamination in many parts of the world: Bangladesh, India, Mexico, parts of the western United States, parts of Eastern Europe) is detoxified through a specific methylation pathway that depends absolutely on SAMe. The enzyme arsenite methyltransferase (AS3MT, formerly Cyt19) sequentially methylates inorganic arsenite (As-III) to methylarsonous acid (MMA-III), then to dimethylarsinous acid (DMA-III), with the methyl groups supplied by SAMe.
The toxicology of arsenic methylation is nuanced. The fully methylated end-product, DMA-V (after oxidation), is relatively non-toxic and is the principal urinary excretion product. However, the intermediate MMA-III is actually more toxic than inorganic arsenite, leading to the paradoxical situation where partial methylation (high MMA, low DMA) may be more harmful than no methylation at all. Population-level studies in arsenic-exposed regions consistently show that individuals with higher DMA:MMA ratio (more complete methylation) have lower rates of arsenic-induced skin lesions, cardiovascular disease, and cancer.
The two principal determinants of arsenic methylation efficiency are:
- AS3MT genetic variation — multiple AS3MT polymorphisms produce slow-methylator and fast-methylator phenotypes. Population distribution varies by ancestry, with high-frequency fast-methylator alleles in some indigenous Andean populations historically exposed to high-arsenic drinking water.
- Methionine and folate status — adequate SAMe supply (which depends on methionine, folate, B12, and B6) supports complete methylation to DMA. Folate supplementation trials in arsenic-exposed populations in Bangladesh have shown that folate repletion shifts the urinary methylation pattern toward more complete methylation (higher DMA:MMA ratio) and is associated with improved biomarkers of arsenic-induced injury.
The clinical implication for arsenic-exposed patients is to ensure adequacy of the entire methionine cycle nutrition: protein adequacy for methionine supply, methylated folate and B12 for the remethylation pathway, B6 for transsulfuration, and selenium (which has been shown in some trials to reduce arsenic toxicity through mechanisms not fully elucidated). The first intervention in any high-exposure setting remains source reduction (alternative water source, point-of-use filtration with arsenic-rated media).
Estrogen Metabolism via COMT
Estrogen metabolism produces a defined sequence of hydroxylated and methylated intermediates, and the clearance of the catecholestrogens (2-hydroxyestrogens and 4-hydroxyestrogens) depends on catechol-O-methyltransferase (COMT), a SAMe-dependent methyltransferase.
The relevant metabolic flow:
- Estradiol and estrone are hydroxylated at the 2 or 4 position by cytochrome P450 enzymes (CYP1A1, CYP1A2, CYP1B1, CYP3A4), producing 2-hydroxyestrogens or 4-hydroxyestrogens. The 2-hydroxy pathway is generally considered the "safer" route and the 4-hydroxy pathway the "riskier" route, because the 4-hydroxy intermediate can be further oxidized to a reactive quinone that forms DNA adducts implicated in estrogen-dependent carcinogenesis.
- The catechol estrogens (both 2- and 4-) are methylated by COMT using SAMe as the methyl donor, producing methoxyestrogens. Methoxyestrogens are non-genotoxic and are excreted via conjugation.
- The competition is between COMT-mediated methylation (the safe clearance route) and further oxidation to quinones (the dangerous route). Adequate SAMe supply and adequate COMT activity favor the safe route.
COMT activity is genetically variable. The Val158Met polymorphism (rs4680) is the most studied: Val/Val homozygotes have approximately 4-fold higher COMT activity than Met/Met homozygotes. The Met/Met phenotype (low COMT activity) is associated with slower estrogen clearance and has been investigated as a potential modifier of breast cancer risk, though the epidemiologic associations are modest and not consistent across populations.
The methionine-pathway implication is that adequate SAMe supply supports estrogen clearance via COMT, and impaired methylation (whether from low methionine intake, severe MTHFR-deficient remethylation, or low B-vitamin cofactor status) may functionally exacerbate the effective Met/Met low-COMT phenotype. In integrative-medicine practice, women with symptoms consistent with estrogen dominance (heavy menstrual bleeding, breast tenderness, fibrocystic breast disease, premenstrual mood symptoms, fibroid uterine pathology) are often supported with methionine-pathway optimization: B-complex with methylfolate and methyl-B12, adequate protein for methionine, optional SAMe trial, optional indole-3-carbinol or diindolylmethane (DIM) to shift CYP1A1/1B1 ratio toward 2-hydroxylation, and adequate fiber for entero-hepatic disruption of estrogen reabsorption.
Histamine Clearance and HNMT
Histamine is cleared by two enzymes operating in parallel:
- Diamine oxidase (DAO) — located primarily in the intestinal mucosa, kidney, and placenta. DAO is the principal enzyme handling dietary histamine and histamine produced by gut microbes. DAO activity is impaired in histamine intolerance, may be reduced by certain medications (NSAIDs, certain antibiotics, alcohol metabolites), and depends on copper and vitamin B6.
- Histamine N-methyltransferase (HNMT) — intracellular cytosolic enzyme present in most tissues, particularly central nervous system, where it is the dominant histamine clearance mechanism. HNMT uses SAMe as the methyl donor.
The clinical syndrome of histamine intolerance, increasingly recognized in integrative-medicine practice, presents with food-related reactions (flushing, headache, urticaria, abdominal symptoms, particularly after high-histamine foods such as aged cheese, fermented foods, red wine, leftovers, smoked or cured meats, certain fish), seasonal allergy patterns, mast cell activation patterns, and sometimes premenstrual exacerbation (estrogen upregulates histamine release from mast cells and downregulates DAO). The most-described clinical pattern in textbooks focuses on DAO insufficiency, but HNMT insufficiency from impaired methylation is a real subset.
The patient who has both DAO insufficiency (often diagnosed by low serum DAO activity or genetic AOC1 polymorphisms) and impaired methylation (elevated homocysteine, low B12, low active folate, severe MTHFR variants) has a double hit on histamine clearance and is particularly prone to histamine intolerance symptoms. Treatment in such cases combines histamine-restricted diet, DAO enzyme supplementation with meals (commercial DAO enzyme products are available, derived from porcine kidney), methylation cofactor support (methylfolate, methyl-B12, P5P), and consideration of SAMe supplementation directly.
Clinical Detoxification Applications
The clinical scenarios where methionine-pathway support is part of a detoxification approach include:
- Documented or suspected heavy metal exposure — mercury (dental amalgam, fish consumption, occupational), lead (old housing, occupational), arsenic (well water), cadmium (smoking, occupational). Methionine-pathway support is adjunctive; specialist toxicology consultation and source reduction are primary.
- Estrogen-dominance symptoms — heavy or painful menstruation, premenstrual mood symptoms, fibrocystic breasts, uterine fibroids, family history of estrogen-dependent cancer. Methionine-pathway support optimizes COMT-mediated estrogen clearance.
- Histamine intolerance — food-triggered symptoms, atopic patterns, mast cell activation patterns. Methionine-pathway support optimizes HNMT-mediated central histamine clearance.
- Recurrent pharmacologic side effects suggesting impaired Phase II conjugation — difficulty tolerating common medications at standard doses, particularly those cleared via glucuronidation or sulfation
- Multiple chemical sensitivity (MCS) — the clinical syndrome of low-threshold reactions to multiple unrelated chemical exposures is heterogeneous and not fully understood, but a subset of patients have demonstrable impaired Phase II conjugation capacity. Methionine-pathway support is reasonable as part of broader environmental medicine evaluation.
- Chronic low-grade inflammatory states with oxidative stress — chronic fatigue syndrome, fibromyalgia, certain autoimmune patterns, post-acute infection syndromes. The methionine-glutathione pathway is part of the antioxidant defense system supporting recovery.
- Pre-surgical and post-surgical optimization — perioperative glutathione is acutely depleted by anesthesia, surgical stress, and inflammatory cytokines. Methionine and NAC support in the weeks before and after major surgery may improve recovery.
- Pre-chemotherapy support (selected cases, with oncology approval) — some chemotherapy regimens deplete glutathione; some are detoxified via glutathione conjugation. NAC and glutathione-supportive nutrition during chemotherapy is controversial and must be coordinated with the oncology team because of theoretical concerns about reducing chemotherapy efficacy.
A typical adult methionine-pathway support stack for detoxification indications includes:
- Methylated B-complex (with L-methylfolate 400-800 mcg and methylcobalamin 500-2500 mcg)
- Pyridoxal-5-phosphate (P5P) 25-50 mg/day
- N-acetylcysteine (NAC) 600 mg 1-2 times daily
- Glycine 2-3 g/day (bone broth or supplemental glycine powder)
- Selenium 100-200 mcg/day (selenomethionine or yeast-derived)
- Zinc 15-30 mg/day
- Magnesium 200-400 mg/day
- Optional: SAMe 400-800 mg/day for specific neuropsychiatric or hepatic indications
- Optional: betaine (TMG) 500-1500 mg/day for elevated homocysteine
- Optional: milk thistle (silymarin) 200-400 mg/day for hepatoprotective effect
Dietary support emphasizes adequate quality protein (eggs, fish, modest amounts of grass-fed meat or quality plant proteins), cruciferous vegetables (broccoli, Brussels sprouts, kale for the indole-3-carbinol effect on estrogen metabolism and the sulforaphane effect on Nrf2 activation), allium vegetables (garlic, onion for sulfur-compound content), and adequate fiber for entero-hepatic disruption.
Cautions in Detoxification Protocols
- Specialist consultation for significant exposures — documented heavy metal exposure with abnormal blood, urine, or hair testing requires consultation with a clinical toxicologist or environmental medicine specialist. Supplement-based home protocols are not appropriate for significant exposures and can mobilize tissue-burden metals in ways that worsen acute symptoms.
- Chelation timing and provocation testing — provoked urine collection (DMSA, DMPS, or EDTA challenge) is sometimes used to estimate tissue heavy-metal burden, but interpretation is complex, and provocation can mobilize metals in clinically relevant amounts. The procedure should be done under specialist supervision with appropriate concurrent renal monitoring and binder support.
- Pregnancy and lactation — do not initiate active heavy-metal detoxification protocols during pregnancy or breastfeeding because mobilized metals can cross to the fetus or appear in breast milk. Methionine-cycle nutrition optimization (adequate protein, methylated B-vitamins, prenatal vitamin) is appropriate and standard, but chelation and aggressive mobilization protocols are not.
- Renal impairment — many detoxification interventions assume normal renal clearance. Patients with reduced GFR (CKD stage 3 or worse) require dose adjustment for NAC, methylcobalamin, and certain mineral supplements, and chelation protocols can transiently worsen renal function.
- Glutathione depletion paradox — aggressive Phase I inducers (alcohol, certain herbal preparations, smoke exposure) without corresponding Phase II support can produce a transient state of high reactive-intermediate burden with inadequate conjugation. The principle in detoxification is to support Phase II before or simultaneously with any intervention that increases Phase I activity, not after.
- Methionine restriction caveat — some longevity-focused practitioners advocate methionine restriction (typically through plant-protein-dominant diets) for healthy adults. This is biologically defensible from the rodent longevity literature but should not be combined with active detoxification protocols that require methionine for glutathione and Phase II support. The two strategies are operating on different timelines and should not be pursued simultaneously.
- Quality of supplemental products — the supplement industry is unevenly regulated. Heavy metal contamination of certain supplements (notably some Ayurvedic preparations, some imported herbal products) has been documented and can contribute to the same exposures the patient is trying to address. Use third-party-tested products from reputable manufacturers (USP, NSF, ConsumerLab certifications).
Key Research Papers
- Stipanuk MH (2004). Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annual Review of Nutrition. — PubMed
- Coles BF, Kadlubar FF (2003). Detoxification of electrophilic compounds by glutathione S-transferase catalysis: determinants of individual response to chemical carcinogens and chemotherapeutic drugs? Biofactors. — PubMed
- Hayes JD, Flanagan JU, Jowsey IR (2005). Glutathione transferases. Annual Review of Pharmacology and Toxicology. — PubMed
- Klaassen CD, Liu J, Diwan BA (2009). Metallothionein protection of cadmium toxicity. Toxicology and Applied Pharmacology. — PubMed
- Bridges CC, Zalups RK (2005). Molecular and ionic mimicry and the transport of toxic metals. Toxicology and Applied Pharmacology. — PubMed
- Vahter M (2002). Mechanisms of arsenic biotransformation. Toxicology. — PubMed
- Gamble MV, Liu X et al. (2006). Folate and arsenic metabolism: a double-blind, placebo-controlled folic acid-supplementation trial in Bangladesh. American Journal of Clinical Nutrition. — PubMed
- Yager JD, Davidson NE (2006). Estrogen carcinogenesis in breast cancer. New England Journal of Medicine. — PubMed
- Tunbridge EM et al. (2006). Catechol-O-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biological Psychiatry. — PubMed
- Maintz L, Novak N (2007). Histamine and histamine intolerance. American Journal of Clinical Nutrition. — PubMed
- Klaassen CD, Reisman SA (2010). Nrf2 the rescue: effects of the antioxidative/electrophilic response on the liver. Toxicology and Applied Pharmacology. — PubMed
- Atmaca G (2004). Antioxidant effects of sulfur-containing amino acids. Yonsei Medical Journal. — PubMed
PubMed Topic Searches
- PubMed: Transsulfuration pathway
- PubMed: Metallothionein and heavy metals
- PubMed: Arsenic methylation
- PubMed: COMT estrogen methylation
- PubMed: HNMT histamine clearance
- PubMed: Sulfation Phase II
Connections
- Methionine Overview
- Methionine Benefits Hub
- Methylation and SAMe
- Methionine for Liver Health
- Methionine for Hair and Nails
- Cysteine
- Taurine
- NAC & Glutathione
- N-Acetylcysteine (NAC)
- Sulfur
- Molybdenum and Detoxification
- Selenium
- Zinc
- Heavy Metals
- Detoxification
- Oxidative Stress
- Homocysteine Lab Test
- All Amino Acids