Methionine for Liver Health
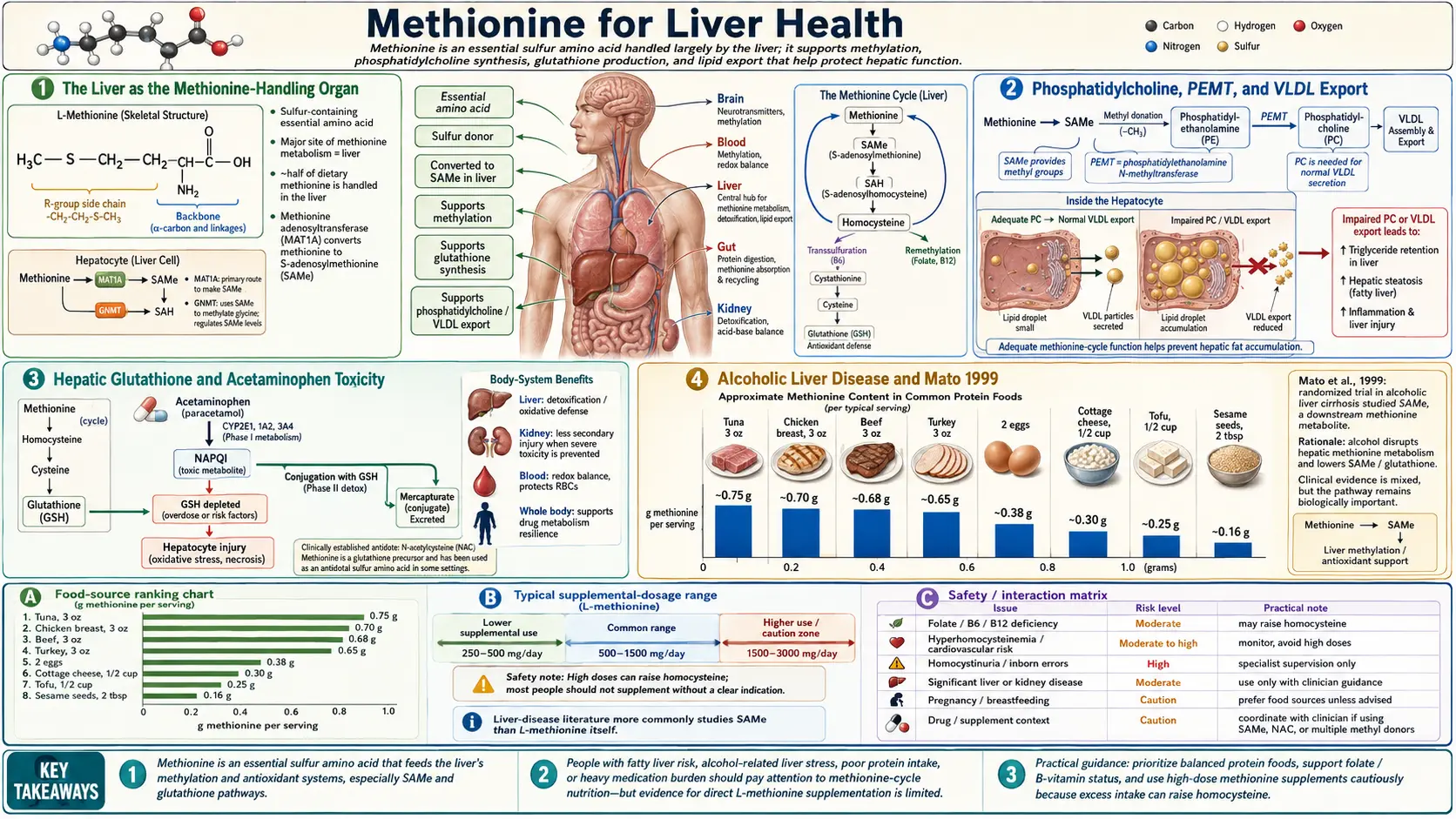
The liver is the single most methionine-dependent organ in the body. Approximately half of all ingested methionine is metabolized in the liver, and hepatic methionine adenosyltransferase (MAT1A, the liver-specific isoform) generates the SAMe used for phosphatidylcholine synthesis, glutathione production, bile acid conjugation, and the methylation of every hormone, drug, and xenobiotic the liver clears. Methionine and SAMe deficiency are central to the pathogenesis of alcoholic liver disease, non-alcoholic fatty liver disease (NAFLD), and intrahepatic cholestasis — and pharmaceutical-grade SAMe has been studied in formal clinical trials for each of these indications. The Mato 1999 trial in alcoholic cirrhosis showed an absolute reduction in death or liver transplant of approximately 12 percentage points in Child-Pugh A and B patients; the European trials in intrahepatic cholestasis of pregnancy show clinically meaningful improvement in pruritus and biochemical markers; the methionine-choline-deficient diet remains the most widely used animal model of non-alcoholic steatohepatitis precisely because removing methionine and choline reliably reproduces the histology of human NASH. This deep-dive walks through each clinical application, the underlying mechanisms, and the practical role of methionine and SAMe in modern hepatology and integrative liver care.
Table of Contents
- The Liver as the Methionine-Handling Organ
- Phosphatidylcholine, PEMT, and VLDL Export
- Hepatic Glutathione and Acetaminophen Toxicity
- Alcoholic Liver Disease and Mato 1999
- The Methionine-Choline-Deficient (MCD) NASH Model
- NAFLD and NASH in Humans
- Intrahepatic Cholestasis of Pregnancy
- Drug-Induced Cholestasis and TPN Cholestasis
- The Methionine Restriction Longevity Counterpoint
- Practical Clinical Application
- Cautions and Drug Interactions
- Key Research Papers
- Connections
- Featured Videos
The Liver as the Methionine-Handling Organ
The liver receives essentially all dietary amino acids via the portal vein after intestinal absorption, and methionine is no exception. Approximately 48% of dietary methionine is metabolized in the first pass through the liver before reaching the systemic circulation, making hepatic methionine handling quantitatively dominant in whole-body methionine economy.
The liver-specific methionine adenosyltransferase isoform (MAT1A) is expressed at much higher levels in adult hepatocytes than the ubiquitous MAT2A isoform expressed elsewhere in the body. MAT1A has lower affinity for methionine but higher capacity, which is appropriate for the high-flux hepatic environment. Mature hepatocytes are SAMe-rich tissue — intracellular SAMe concentrations of 50-100 micromolar are routine, compared to 5-50 micromolar in most other tissues.
This dense hepatic SAMe pool serves three major hepatic functions:
- Phosphatidylcholine synthesis via PEMT for VLDL packaging and bile membrane integrity
- Glutathione synthesis via the transsulfuration pathway (cysteine substrate supply)
- Methylation of xenobiotics, hormones, and bile acids for their inactivation and excretion
Loss of hepatic MAT1A activity (whether from chronic ethanol consumption, severe liver disease, or experimental knockout in animals) produces a recognizable hepatic phenotype: SAMe depletion, glutathione depletion, fatty liver, hepatocyte apoptosis, oxidative stress, and predisposition to fibrosis and hepatocellular carcinoma. The MAT1A-knockout mouse develops spontaneous fatty liver and HCC, providing genetic confirmation that methionine flux through the SAMe pool is hepatoprotective.
Phosphatidylcholine, PEMT, and VLDL Export
One of the liver's defining metabolic responsibilities is to package triglycerides into very-low-density lipoprotein (VLDL) particles for export to peripheral tissues. VLDL assembly requires phosphatidylcholine (PC) as the outer monolayer phospholipid of the lipoprotein particle. When PC is in short supply, VLDL assembly stalls, triglycerides accumulate within the hepatocyte cytoplasm as lipid droplets, and the histologic picture of hepatic steatosis develops.
The liver produces PC through two pathways:
- The CDP-choline pathway (Kennedy pathway) uses dietary choline. Choline is phosphorylated, activated to CDP-choline, and conjugated to diacylglycerol.
- The PEMT pathway (phosphatidylethanolamine N-methyltransferase) sequentially methylates phosphatidylethanolamine three times using SAMe as the methyl donor. PEMT is uniquely expressed at high levels in hepatocytes (it is the only mammalian methyltransferase that uses a phospholipid substrate) and produces approximately 30% of hepatic PC under typical dietary conditions.
The interconvertibility of these two pathways is critical. When dietary choline is adequate, the CDP-choline pathway dominates and the PEMT pathway is downregulated. When dietary choline is restricted, the PEMT pathway must scale up, increasing methionine demand for the SAMe consumed in the three methylation steps. When both methionine and choline are restricted simultaneously, neither pathway can produce adequate PC, VLDL assembly fails, and fatty liver develops rapidly — the mechanistic basis for the methionine-choline-deficient diet model discussed below.
PEMT-derived PC has a different fatty acid composition than CDP-choline-derived PC — specifically, PEMT-PC is enriched in long-chain polyunsaturated fatty acids (especially arachidonic acid and DHA) in the sn-2 position. This compositional difference matters for VLDL packaging because the most actively secreted VLDL particles preferentially incorporate PEMT-PC. The PEMT-knockout mouse develops steatohepatitis on a choline-deficient background; the mouse cannot compensate for choline deficiency through PEMT and accumulates hepatic triglycerides.
Human PEMT polymorphisms have been documented and are associated with risk of NAFLD in choline-marginal diets, particularly in postmenopausal women whose estrogen-mediated PEMT induction has fallen. The clinical implication is that adequate methionine intake helps protect against choline-marginal dietary patterns by maintaining functional PEMT activity.
Hepatic Glutathione and Acetaminophen Toxicity
Hepatic glutathione concentration is among the highest of any tissue in the body, reaching 5-10 millimolar in healthy hepatocytes — an order of magnitude above most other cell types. This high concentration is necessary because the liver is the primary site of xenobiotic metabolism, and glutathione conjugation is the dominant Phase II detoxification reaction for reactive electrophiles produced by Phase I cytochrome P450 oxidation.
The rate-limiting precursor for glutathione synthesis is cysteine, which the hepatocyte produces either by transsulfuration from methionine (the pathway described in our Methylation and SAMe deep-dive) or by direct uptake from circulation. Under conditions of methionine limitation, the transsulfuration pathway is impaired and hepatic glutathione synthesis is constrained.
The clinical relevance of this mechanism is most dramatic in acetaminophen (paracetamol) toxicity. Therapeutic acetaminophen doses are metabolized primarily by glucuronidation (Phase II) with a small fraction (5-15%) flowing through CYP2E1 to produce N-acetyl-p-benzoquinone imine (NAPQI), a reactive electrophile that is normally inactivated by glutathione conjugation. At supratherapeutic doses, NAPQI production overwhelms glutathione supply, free NAPQI binds covalently to hepatocyte proteins, and centrilobular hepatic necrosis ensues. This is the most common cause of acute liver failure in the United States.
The antidote, N-acetylcysteine (NAC), works by directly supplying cysteine for emergency glutathione synthesis. NAC is the cysteine donor of choice for this purpose because it has better oral bioavailability than free cysteine and is not subject to first-pass hepatic catabolism. In any chronic clinical setting that depletes hepatic glutathione — ongoing acetaminophen use at the upper end of dosing, alcoholic liver disease, NAFLD, certain chemotherapy regimens — adequate dietary methionine plus supplemental NAC (typical dose 600-1800 mg/day) provides combined upstream and direct substrate support for glutathione synthesis.
For the full discussion of glutathione and NAC, see our NAC & Glutathione page.
Alcoholic Liver Disease and Mato 1999
Chronic ethanol consumption causes profound disturbances of hepatic methionine and SAMe metabolism. The relevant mechanisms include:
- Reduced MAT1A activity — ethanol downregulates MAT1A expression and post-translationally inhibits the enzyme, reducing SAMe synthesis from methionine.
- Elevated SAH — ethanol metabolism produces acetaldehyde, which inhibits S-adenosylhomocysteine hydrolase (SAHH). SAH accumulates, the SAH/SAMe ratio rises, and the cellular methylation potential falls.
- Glutathione depletion — combined methionine flux impairment and direct oxidative stress from ethanol metabolism deplete hepatic glutathione.
- Mitochondrial dysfunction — the liver mitochondrial glutathione pool is particularly vulnerable because mitochondrial glutathione import depends on functional inner membrane carriers that are impaired in alcoholic liver disease.
- Reduced phosphatidylcholine — impaired PEMT activity (SAMe-dependent) plus reduced dietary choline contribute to hepatic steatosis and impaired membrane fluidity.
The clinical translation came in the form of the Mato 1999 multicenter European trial. Mato and colleagues randomized 123 patients with alcoholic cirrhosis (Child-Pugh A, B, or C) to oral SAMe 1200 mg/day or placebo for 24 months. The primary endpoint was death or liver transplantation. Results:
- Overall mortality or transplant: SAMe 16% vs placebo 30% (p=0.077 in the full intent-to-treat analysis)
- In the Child-Pugh A and B subgroup (excluding the most severe C patients): SAMe 12% vs placebo 29% (p=0.025)
- No significant safety signal
The Mato trial established SAMe (Italian brand name Samyr, intravenous and oral; US OTC since 1999) as a clinically meaningful intervention for alcoholic liver disease. The effect size in the Child-Pugh A and B subgroup — an absolute mortality reduction of 17 percentage points over 24 months — would be considered substantial for any liver disease intervention. The trial has been criticized for the underpowering of the full ITT analysis, but the biological rationale (correcting the documented hepatic SAMe deficit) is strong and the safety profile is benign, so SAMe has been adopted into integrative-medicine and hepatology practice for alcoholic liver disease management.
Subsequent meta-analyses (the Cochrane review and Lieber narrative reviews) have largely confirmed a clinical benefit in alcoholic liver disease without firmly establishing optimal dose, formulation, or duration. The European Association for the Study of the Liver (EASL) guidelines acknowledge SAMe as a reasonable adjunct in selected patients with alcoholic liver disease.
The Methionine-Choline-Deficient (MCD) NASH Model
The methionine-choline-deficient (MCD) diet is the most widely used and most reproducible rodent dietary model of non-alcoholic steatohepatitis (NASH). Feeding mice or rats a chow that lacks both methionine and choline produces, within 2-4 weeks, the full histologic spectrum of human NASH:
- Macrovesicular and microvesicular steatosis (lipid droplet accumulation in hepatocytes)
- Hepatocyte ballooning
- Lobular inflammation with neutrophil and lymphocyte infiltration
- Mallory-Denk bodies
- Pericellular and perisinusoidal fibrosis with prolonged feeding (8-12 weeks)
The mechanism is the convergent failure of both phosphatidylcholine synthesis pathways. Without methionine, the SAMe supply for PEMT is exhausted within days. Without choline, the CDP-choline pathway has no substrate. With both pathways inoperative, hepatic PC drops, VLDL assembly fails, triglycerides accumulate, and the secondary cascade of lipotoxicity, oxidative stress, and inflammation produces full-blown steatohepatitis.
The MCD model has the limitation that animals lose weight rather than gain it (the opposite of human NAFLD, which is typically associated with obesity and insulin resistance). This has led to refinements such as the choline-deficient L-amino-acid-defined (CDAA) diet (which provides methionine but eliminates choline and uses purified amino acids), the high-fat MCD variant, and the "Western diet" with reduced choline. All produce variations on the same hepatic phenotype.
The translational message from MCD is unambiguous: simultaneously inadequate methionine and choline intake produces the histologic hallmarks of NASH. Whether marginal dietary insufficiency of methionine and choline contributes to human NAFLD in the typical clinical setting is the more nuanced question, addressed in the next section.
NAFLD and NASH in Humans
Non-alcoholic fatty liver disease (NAFLD) is now the most prevalent chronic liver disease worldwide, affecting approximately 25-30% of adults in developed countries. A subset of NAFLD patients progress to non-alcoholic steatohepatitis (NASH) with active inflammation and risk of fibrosis, cirrhosis, and hepatocellular carcinoma. The principal drivers in humans are obesity, insulin resistance, type 2 diabetes, dyslipidemia, and high-fructose dietary patterns — not primarily methionine or choline deficiency.
However, the methionine and choline pathway intersects with human NAFLD/NASH in several clinically relevant ways:
- Choline inadequacy — the dietary reference intake for choline (550 mg/day for men, 425 mg/day for women) is not met by approximately 90% of US adults per NHANES data. Postmenopausal women, on whom estrogen-mediated PEMT induction is no longer available, are particularly vulnerable. Adequate methionine intake helps the liver compensate for marginal choline intake by maintaining PEMT capacity.
- PEMT polymorphisms — the PEMT rs7946 polymorphism (V175M) reduces enzyme activity and is associated with higher risk of NAFLD on choline-marginal diets. Such patients may benefit from higher dietary choline intake.
- SAMe trials in NAFLD/NASH — smaller human trials of SAMe in NAFLD have generally shown improvement in transaminases and some imaging-based fibrosis markers but have not been definitive. The most rigorous SAMe-for-NAFLD trials are still in progress. The pre-existing approval of SAMe as OTC supplement complicates traditional pharmaceutical trial economics.
- Phosphatidylcholine supplementation — oral PC (lecithin-derived, soybean or sunflower-source) is sometimes used as adjunct in NAFLD with mixed evidence. Polyenylphosphatidylcholine (PPC, the Essentiale formulation widely used in Russia, Germany, and Eastern Europe) has the longest clinical track record, primarily for alcoholic and toxic liver injury.
- Betaine supplementation — betaine (trimethylglycine, TMG) provides the alternative BHMT remethylation pathway for homocysteine in liver and kidney. Small NAFLD trials have used betaine 10-20 g/day with modest improvements in transaminases and imaging steatosis scores.
For clinical NAFLD/NASH management, the primary interventions remain weight loss, glycemic control, and treatment of associated metabolic syndrome features. Adequate dietary methionine and choline are supportive but are not standalone treatments for established disease. Patients on highly restricted vegan diets are at structural risk for both methionine inadequacy and choline inadequacy and warrant nutritional assessment in any liver-disease context.
Intrahepatic Cholestasis of Pregnancy
Intrahepatic cholestasis of pregnancy (ICP) is a reversible cholestatic disorder that develops in 0.5-2% of pregnancies, most commonly in the third trimester. It is characterized by intense maternal pruritus (particularly palmar and plantar), elevated serum bile acids, modest elevation of transaminases, and (in severe cases) increased fetal risk of preterm birth, meconium-stained amniotic fluid, and stillbirth. The condition resolves rapidly postpartum but recurs in approximately 60-70% of subsequent pregnancies.
The mechanism involves estrogen-induced impairment of hepatocyte canalicular bile acid transport (BSEP, the bile salt export pump, encoded by ABCB11) and impaired phospholipid efflux (MDR3, encoded by ABCB4). Genetic variants in BSEP and MDR3 predispose to recurrent ICP and to similar but more severe pediatric cholestatic syndromes (PFIC types 1-3).
First-line treatment is ursodeoxycholic acid (UDCA, ursodiol) at 10-15 mg/kg/day, which displaces hydrophobic bile acids and improves pruritus and fetal outcomes. SAMe has been studied as monotherapy and as add-on to UDCA in multiple controlled trials, with European trials (particularly Ribalta 1991 and several subsequent Italian studies) showing significant improvement in pruritus, serum bile acids, and biochemical markers. The Cochrane review concludes that UDCA is superior to SAMe as monotherapy, but combination UDCA plus SAMe is more effective than UDCA alone for severe or refractory cases. SAMe is typically given at 800-1000 mg/day orally (intravenous SAMe is used in severe cases in hospital settings).
The mechanistic rationale is that SAMe supports hepatic phosphatidylcholine synthesis (membrane fluidity required for canalicular function), supports hepatic glutathione (membrane protection from oxidative stress during cholestasis), and provides methyl groups for the synthesis of methylated bile acids that are less hepatotoxic than unconjugated species. Combined with UDCA's hydrophobic-bile-acid displacement, the two interventions are synergistic.
Drug-Induced Cholestasis and TPN Cholestasis
SAMe has been studied in two additional cholestatic indications:
- Drug-induced cholestasis — multiple medications (estrogens, anabolic steroids, oral contraceptives, certain antibiotics, ciclosporin) cause cholestatic liver injury through mechanisms that overlap with the ICP picture. Small SAMe trials have shown biochemical improvement in patients with cyclosporin-induced cholestasis after organ transplantation and in patients with estrogen-induced cholestasis. The clinical utility is most established in the post-transplant ciclosporin setting where the alternative is dose reduction or drug withdrawal, both of which compromise immunosuppression.
- Total parenteral nutrition (TPN) cholestasis — long-term TPN, particularly in pediatric patients on intestinal failure regimens, causes a progressive cholestatic liver injury that historically progressed to cirrhosis and need for combined liver-intestine transplantation. SAMe has been used as adjunct in TPN-associated liver disease with the rationale that bypassing the GI tract bypasses the normal methionine-cycle bile-acid flux. Evidence is preliminary; more impactful interventions have been the switch from soy-based to fish-oil-based intravenous lipid emulsions and intestinal rehabilitation programs.
The Methionine Restriction Longevity Counterpoint
An honest discussion of methionine and hepatic health must address the apparently paradoxical observation that, in laboratory rodents, dietary methionine restriction (typically 0.17% methionine vs 0.86% control) extends maximum lifespan by 30-45% in multiple independent studies, with some life-extending benefit even when started in middle age. This is among the most robust dietary longevity interventions known in mammalian models.
The mechanism appears to involve:
- Improved hepatic insulin sensitivity — methionine restriction rapidly improves hepatic insulin signaling, reduces hepatic glucose output, and improves whole-body glucose tolerance even in obese mice.
- Reduced hepatic mitochondrial ROS production — methionine-restricted mitochondria produce less hydrogen peroxide at complex I, the principal site of age-related mitochondrial oxidative stress.
- Activation of FGF21 secretion — the liver responds to amino acid restriction (particularly methionine restriction) by secreting FGF21, a metabolic hormone that promotes fat oxidation, improves insulin sensitivity, and may underlie some of the longevity effect.
- Reduced mTORC1 signaling — methionine and leucine are the two strongest activators of the mTORC1 nutrient-sensing complex. Methionine restriction reduces mTORC1 activity, mimicking some of the effects of caloric restriction or rapamycin.
- Changed transsulfuration flux — methionine restriction shifts the methionine cycle equilibrium and changes the SAH/SAMe ratio in ways that produce widespread epigenetic remodeling.
Whether methionine restriction extends healthspan in humans is unknown. Several small short-term human trials of methionine restriction (typically achieved through plant-protein-dominant or whey-isolate vegan diets) show metabolic improvements: reduced fasting insulin, lower fasting glucose, improved lipid profile, lower plasma methionine and homocysteine, and increased FGF21. The trials have not been long enough or large enough to detect lifespan or cardiovascular endpoints.
The clinical implication for hepatic disease is nuanced. Methionine and SAMe are unambiguously hepatoprotective in the setting of an already-injured liver (alcoholic, cholestatic, NAFLD with established steatohepatitis), and clinical trials have established the role of SAMe supplementation in these conditions. By contrast, in the otherwise healthy adult on a high-protein omnivorous diet, the longevity literature raises the question of whether chronic methionine excess (typical in modern Western diets, often 2-3 grams per day from animal-protein-heavy patterns) may be contributing to age-related metabolic decline. The honest answer is that we do not yet know with confidence, but the question is real, and patients considering very-high-protein interventions (carnivore diets, ultra-high-protein bodybuilding regimens) should be aware that the longevity rodent literature suggests caution.
A practical middle ground that respects both the hepatoprotective and the longevity literatures is moderate dietary methionine (typical mixed-diet intake of 1.0-1.5 g/day for a 70 kg adult) with adequate B-vitamin cofactor support, plus targeted SAMe supplementation only when there is a specific clinical indication.
Practical Clinical Application
For patients with active liver disease, the integrative-medicine approach combines methionine-pathway support with the underlying-disease management. Specific scenarios:
- Alcoholic liver disease (active or recent) — abstinence is non-negotiable. On top of abstinence, consider SAMe 800-1200 mg/day (enteric-coated, divided dosing), thiamine 100 mg/day, folate 1 mg/day, NAC 600-1200 mg/day, and zinc 30 mg/day. Monitor LFTs, INR, albumin, and bilirubin. Specialist hepatology referral for Child-Pugh B or C cirrhosis.
- NAFLD without significant fibrosis — weight loss is the primary intervention. Adjuncts: ensure choline adequacy (eggs, organ meats, soy, or supplemental choline bitartrate 500-1000 mg/day in choline-restricted diets), adequate methionine via protein intake, optional SAMe 400-800 mg/day, optional betaine 1000-3000 mg/day, NAC 600 mg/day. Vitamin E 400-800 IU/day has the strongest evidence base for NASH in non-diabetic patients.
- NASH with fibrosis — specialist hepatology and consideration of pharmacologic agents (pioglitazone for biopsy-confirmed NASH in selected patients; resmetirom and other newer agents). The supplement stack above remains supportive but is not a substitute for weight management and pharmacologic therapy where indicated.
- Intrahepatic cholestasis of pregnancy — UDCA 10-15 mg/kg/day is first-line. SAMe 800-1000 mg/day may be added for severe or refractory cases under obstetric supervision. Always involve obstetric medicine.
- Acetaminophen exposure or chronic high-dose use — NAC 600 mg twice daily for chronic use; NAC by emergency-department protocol for acute overdose (within 8 hours of ingestion is optimal but can be effective up to 24-48 hours).
- Drug-induced hepatocellular injury — discontinue offending agent. Supportive care. NAC and SAMe may be considered.
- Chronic methionine pathway support (otherwise healthy patient with elevated homocysteine) — methylated B-complex with methylfolate and methylcobalamin, B6 as P5P, betaine 500-1500 mg/day if homocysteine remains elevated. Recheck homocysteine at 8-12 weeks.
For the related discussion of liver detoxification and conjugation pathways, see our Methionine for Detoxification deep-dive.
Cautions and Drug Interactions
- Active liver failure — in severe acute liver failure (INR >1.5, encephalopathy), management is hospital-based and includes consideration of liver transplant. Supplement-based methionine-pathway support is adjunctive and never substitutes for specialist hepatology and transplant evaluation.
- SAMe and bipolar disorder — like any antidepressant, SAMe can precipitate or worsen mania. Screen for personal or family history of bipolar disorder before initiating, particularly in patients receiving SAMe for liver indications who may not be aware of its antidepressant activity.
- Methionine loading and homocysteine — very-high-dose methionine (such as the "methionine loading test" once used to diagnose CBS deficiency) acutely raises homocysteine and is contraindicated in patients with severe vascular disease, recent stroke, or known severe CBS deficiency.
- Cysteine-derived hydrogen sulfide accumulation — in cystathionine gamma-lyase (CGL) deficiency or in some MTHFR/CBS combinations, methionine flux can accumulate as cysteine and hydrogen sulfide. Rare scenario but specialist metabolic disease territory.
- Drug interactions — SAMe with SSRIs/SNRIs/MAOIs may theoretically increase serotonin syndrome risk; the combination is commonly used clinically (Papakostas augmentation paradigm) but warrants monitoring. SAMe may reduce levodopa efficacy through peripheral COMT induction. NAC can mildly potentiate nitrate vasodilators.
- Lipotropic combination products — classical lipotropic formulas combining methionine, choline, inositol, and B-vitamins are widely available. Quality varies. Patients on such formulas should be screened for active liver disease that warrants formal evaluation rather than supplement-based self-management.
Key Research Papers
- Mato JM et al. (1999). S-adenosylmethionine in alcoholic liver cirrhosis: a randomized, placebo-controlled, double-blind, multicenter clinical trial. Journal of Hepatology. — PubMed
- Lu SC, Mato JM (2012). S-adenosylmethionine in liver health, injury, and cancer. Physiological Reviews. — PubMed
- Mato JM, Martínez-Chantar ML, Lu SC (2008). Methionine metabolism and liver disease. Annual Review of Nutrition. — PubMed
- Vance DE (2014). Phospholipid methylation by PEMT: unexpected links to non-alcoholic fatty liver disease and steatohepatitis. Biochemistry and Cell Biology. — PubMed
- Anstee QM, Goldin RD (2006). Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. International Journal of Experimental Pathology (MCD diet review). — PubMed
- Ribalta J et al. (1991). S-adenosyl-L-methionine in the treatment of patients with intrahepatic cholestasis of pregnancy. Hepatology. — PubMed
- Zhang L et al. (2017). Ursodeoxycholic acid and S-adenosylmethionine in intrahepatic cholestasis of pregnancy: a network meta-analysis. Hepatology Research. — PubMed
- Anstee QM, Day CP (2012). S-adenosylmethionine (SAMe) therapy in liver disease: a review of current evidence and clinical utility. Journal of Hepatology. — PubMed
- Orentreich N et al. (1993). Low methionine ingestion by rats extends life span. Journal of Nutrition. — PubMed
- Miller RA et al. (2005). Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels. Aging Cell. — PubMed
- Lieber CS (2002). S-adenosyl-L-methionine: its role in the treatment of liver disorders. American Journal of Clinical Nutrition. — PubMed
- Zeisel SH (2006). Choline: critical role during fetal development and dietary requirements in adults. Annual Review of Nutrition. — PubMed
PubMed Topic Searches
- PubMed: SAMe for alcoholic liver disease
- PubMed: MCD diet NASH model
- PubMed: PEMT and fatty liver
- PubMed: ICP and SAMe
- PubMed: Methionine restriction longevity
Connections
- Methionine Overview
- Methionine Benefits Hub
- Methylation and SAMe
- Methionine for Detoxification
- Methionine for Hair and Nails
- Cysteine
- NAC & Glutathione
- N-Acetylcysteine (NAC)
- Homocysteine Lab Test
- Fatty Liver Disease
- Vitamin B12
- Vitamin B6
- Heavy Metals
- Organ Meats (Liver)
- Eggs (Choline + Methionine)
- All Amino Acids
- Liver Disease — the general overview of hepatic disease, the clinical context for methionine and SAMe support.