Methionine for Methylation and SAMe
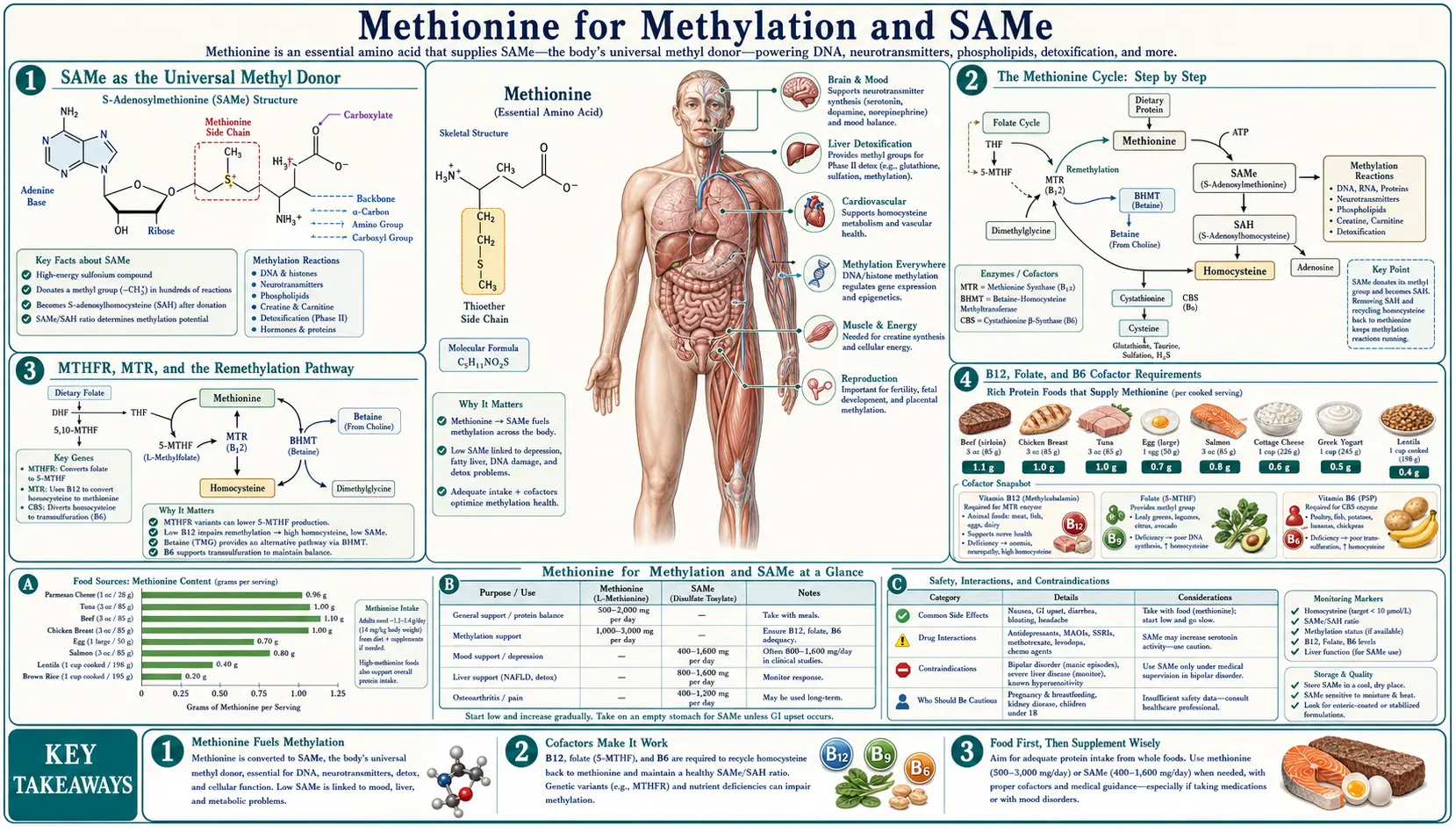
Methionine's most consequential biochemical role is to serve as the metabolic source of S-adenosylmethionine (SAMe), the universal methyl donor that drives more than 200 distinct enzymatic reactions in the human body. SAMe-dependent methylation regulates gene expression through DNA methylation, modulates chromatin via histone methylation, synthesizes the neurotransmitters serotonin, dopamine, melatonin, epinephrine, and norepinephrine, builds the phospholipid phosphatidylcholine for every cell membrane, manufactures creatine and carnitine for energy metabolism, and inactivates excess hormones, drugs, and histamine. The full methionine cycle — methionine to SAMe to SAH to homocysteine and then back to methionine via folate/B12 or forward to cysteine via B6 — sits at the center of one-carbon metabolism, the integrating biochemical hub that links amino acid nutrition, B-vitamin status, MTHFR genetics, neurotransmitter biosynthesis, cardiovascular risk, and detoxification capacity. This deep-dive walks through the mechanism, the enzymes, the cofactor requirements, the clinical SAMe trials, and what the methylation paradigm means for adult patients managing mood disorders, MTHFR polymorphisms, or chronic methylation impairment.
Table of Contents
- SAMe as the Universal Methyl Donor
- The Methionine Cycle: Step by Step
- MTHFR, MTR, and the Remethylation Pathway
- B12, Folate, and B6 Cofactor Requirements
- DNA and Histone Methylation (Epigenetics)
- Neurotransmitter Synthesis via SAMe
- Phosphatidylcholine and Membrane Synthesis
- SAMe Clinical Trials (Depression, Liver, Osteoarthritis)
- Methylation Impairment: Clinical Pattern
- Practical SAMe and Methionine Supplementation
- Cautions and Drug Interactions
- Key Research Papers
- Connections
- Featured Videos
SAMe as the Universal Methyl Donor
S-adenosylmethionine (SAMe, also written SAM, AdoMet, or S-adenosyl-L-methionine) was first isolated by Giulio Cantoni in 1952 at the National Institutes of Health. Cantoni recognized that the molecule possessed a high-energy sulfonium center that made its bound methyl group remarkably reactive — far more reactive than the methyl group on methionine itself or on choline. The activation step from methionine to SAMe consumes the entire triphosphate energy of one ATP molecule (yielding inorganic phosphate plus pyrophosphate), which is why methylation is one of the most energetically expensive routine biochemical operations in the cell.
The reason this energy investment is biologically worthwhile is that SAMe is the only practical donor for the vast majority of biological methyl transfers. More than 200 distinct methyltransferase enzymes in the human genome rely on SAMe as their methyl source. The list of products covers a strikingly broad swath of cellular function:
- Genome regulation — DNA methylation at cytosine residues of CpG dinucleotides (DNMT1, DNMT3A, DNMT3B enzymes) and histone methylation at lysine and arginine residues (more than 50 histone methyltransferases). Together these constitute the epigenome — the layer of chemical marks superimposed on DNA that determines which genes are expressed in which tissues.
- Neurotransmitter synthesis — phenylethanolamine N-methyltransferase (PNMT) converts norepinephrine to epinephrine. Catechol-O-methyltransferase (COMT) inactivates dopamine, norepinephrine, and epinephrine. The melatonin synthesis pathway requires SAMe at the final step (hydroxyindole-O-methyltransferase). Serotonin precursor metabolism depends on methylation, and the synthesis of all biogenic amines is methylation-coupled.
- Phospholipid synthesis — the PEMT enzyme (phosphatidylethanolamine N-methyltransferase) converts phosphatidylethanolamine to phosphatidylcholine through three sequential SAMe-dependent methylations. This is the alternative pathway to dietary choline for phosphatidylcholine production and is dominant in the liver.
- Energy metabolism — creatine synthesis (guanidinoacetate N-methyltransferase, GAMT) consumes approximately 75% of all daily SAMe-derived methyl groups in the adult human. Carnitine synthesis also requires methylated lysine residues in the precursor protein.
- Detoxification and hormone clearance — SAMe-dependent methylation inactivates histamine (HNMT), inactivates estrogens (COMT methylates 2-hydroxyestradiol and 4-hydroxyestradiol), inactivates certain xenobiotics, and methylates arsenic for excretion (AS3MT).
- Capping of messenger RNA — the 5' cap of every messenger RNA molecule includes a SAMe-derived methyl group required for ribosomal recognition and stability.
This functional breadth is the reason a single biochemical bottleneck in methionine or SAMe availability produces clinical pictures that touch every organ system — depression and cognitive dysfunction (neurotransmitter deficit), fatty liver (impaired PEMT-dependent phosphatidylcholine synthesis), muscle weakness (impaired creatine synthesis), abnormal epigenetic programming (impaired DNA methylation), elevated histamine and estrogen burden (impaired clearance), and cardiovascular risk (homocysteine accumulation as discussed below).
The Methionine Cycle: Step by Step
The methionine cycle is the central biochemical loop that interconverts methionine and homocysteine through SAMe, and it represents the operational core of one-carbon metabolism. Walking through it step by step:
- Methionine plus ATP to SAMe (enzyme: methionine adenosyltransferase, MAT2A in most tissues, MAT1A in hepatocytes). This is the activation step that converts the relatively unreactive methyl group on methionine into the high-energy methyl group on SAMe by appending the adenosyl group of ATP. The reaction consumes all three phosphates of ATP, releasing both pyrophosphate and inorganic phosphate — an unusual energetic profile for an ATP-using reaction.
- SAMe donates its methyl group to a substrate (enzyme: any of more than 200 methyltransferases). The methyl group is transferred to the methyl-accepting substrate (DNA, histone, neurotransmitter, phospholipid, etc.), leaving behind S-adenosylhomocysteine (SAH).
- SAH to homocysteine plus adenosine (enzyme: S-adenosylhomocysteine hydrolase, SAHH or AHCY). This hydrolytic step is reversible and is product-inhibited — if adenosine or homocysteine accumulates, the equilibrium pushes back toward SAH, which then accumulates and inhibits the methyltransferases of the previous step. This product-inhibition feedback loop is why an elevated SAH/SAMe ratio is the truest biochemical signature of methylation impairment.
- Homocysteine has three fates:
- Remethylation back to methionine via folate-dependent pathway (enzyme: methionine synthase, MTR) — uses 5-methyltetrahydrofolate as the methyl source and vitamin B12 (methylcobalamin) as the obligate cofactor. This pathway closes the methionine cycle and recycles the molecule for another round of methyl donation.
- Remethylation back to methionine via betaine-dependent pathway (enzyme: betaine-homocysteine methyltransferase, BHMT) — uses betaine (trimethylglycine, TMG) as the methyl source. This pathway is hepatic and renal and does not require folate or B12. It serves as a backup pathway, particularly important in fatty liver disease, alcoholic liver disease, and inherited MTHFR enzyme deficits.
- Transsulfuration to cysteine (enzymes: cystathionine beta-synthase, CBS, then cystathionine gamma-lyase, CGL) — commits the sulfur of homocysteine to cysteine production, which then flows into glutathione synthesis, taurine synthesis, and the body's sulfur reserve. CBS requires vitamin B6 (pyridoxal-5-phosphate) and is activated by SAMe — when SAMe levels are high, CBS is activated and shunts homocysteine toward cysteine; when SAMe is low, CBS is inactive and homocysteine is preferentially recycled back to methionine.
The branching at homocysteine between the remethylation pathway (folate/B12 or betaine) and the transsulfuration pathway (B6) is one of the most clinically consequential decision points in human biochemistry. It determines whether the body conserves methionine for continued methylation (recycling) or commits the sulfur to glutathione and cysteine for antioxidant/detoxification work (one-way transsulfuration). The SAMe-mediated regulation of CBS ensures that methylation needs are met first, and only when methionine is abundant does excess flow toward cysteine.
For the related transsulfuration discussion focused on glutathione synthesis, see our NAC and Glutathione page and our Cysteine page.
MTHFR, MTR, and the Remethylation Pathway
The most clinically discussed component of the methionine cycle in the past two decades has been the MTHFR gene and its common polymorphisms. MTHFR (methylenetetrahydrofolate reductase) does not directly handle methionine or homocysteine — it operates upstream, in the folate cycle, where it converts 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate. The 5-MTHF product is then handed off to MTR (methionine synthase) which uses it to remethylate homocysteine back to methionine.
Two common single-nucleotide polymorphisms reduce MTHFR enzyme activity:
- MTHFR C677T (rs1801133) — the most studied variant. Heterozygotes (CT) retain approximately 65% of wild-type enzyme activity. Homozygotes (TT) retain only about 30% of wild-type activity. The allele frequency varies dramatically by population: roughly 10-15% TT in Caucasian and East Asian populations, lower in African populations.
- MTHFR A1298C (rs1801131) — less impactful in isolation but compound heterozygotes (677CT plus 1298AC) functionally resemble 677TT homozygotes and show similarly elevated homocysteine.
The downstream consequence of reduced MTHFR activity is reduced 5-MTHF supply to methionine synthase, leading to reduced remethylation of homocysteine and (in the setting of marginal folate or B12 intake) modest elevations in fasting plasma homocysteine. The size of this effect is typically 2-3 micromoles/L, which is biologically small but cumulatively meaningful as a cardiovascular risk factor over decades.
MTR (methionine synthase) itself has clinically relevant polymorphisms (A2756G, rs1805087), and its essential cofactor methionine synthase reductase (MTRR, A66G rs1801394) likewise has variants that modestly impair enzyme function. The MTRR enzyme regenerates the active methylcobalamin form of vitamin B12 after each methylation cycle — without it, the B12 on MTR becomes oxidized and the enzyme stalls.
The practical clinical bottom line is that MTHFR polymorphisms are common, biochemically real, and adequately compensated by dietary folate and B12 sufficiency in most patients. The patient who genuinely benefits from clinical attention to MTHFR genotype is the one with documented elevated fasting homocysteine (above approximately 10 micromoles/L), particularly if combined with cardiovascular disease, recurrent pregnancy loss, or a treatment-resistant depression that has not responded to conventional antidepressants. For those patients, supplementation with the activated cofactors — methylfolate (L-5-MTHF), methylcobalamin (methyl B12), and pyridoxal-5-phosphate (active B6) — bypasses the impaired MTHFR step and typically normalizes homocysteine within 8-12 weeks.
For the full clinical workup of elevated homocysteine, see our Homocysteine lab test page.
B12, Folate, and B6 Cofactor Requirements
The methionine cycle and the parallel folate cycle have an absolute, non-substitutable requirement for three B vitamin cofactors. Deficiency of any one of them throws the entire one-carbon metabolism system into disarray:
- Vitamin B12 (cobalamin) — required by methionine synthase (MTR) to receive the methyl group from 5-MTHF and transfer it to homocysteine. B12 also serves as cofactor for methylmalonyl-CoA mutase in a separate pathway (propionate metabolism), so B12 deficiency produces both elevated homocysteine and elevated methylmalonic acid (MMA). The MMA elevation is the more specific marker of B12 deficiency because folate deficiency only elevates homocysteine. B12 absorption requires intact gastric intrinsic factor; pernicious anemia, atrophic gastritis, long-term proton pump inhibitor use, metformin therapy, and bariatric surgery all impair absorption. Vegetarians and vegans are at structural risk because B12 is essentially absent from plant foods other than fortified products.
- Folate (vitamin B9) — required as 5-methyltetrahydrofolate to donate the methyl group used by methionine synthase. Folate also has many functions beyond the methionine cycle, including thymidylate synthesis (DNA precursor) and purine synthesis. The folate cycle is functionally coupled to the methionine cycle — an impaired methionine cycle creates the "methyl trap" in which folate becomes trapped as 5-MTHF and cannot regenerate the other folate species needed for nucleotide synthesis. This is the mechanism behind megaloblastic anemia in B12 deficiency (where folate appears adequate but is functionally trapped). Activated dietary folate (L-5-MTHF) bypasses both the MTHFR step and the methyl-trap problem.
- Vitamin B6 (pyridoxal-5-phosphate) — required by cystathionine beta-synthase (CBS) to commit homocysteine to the transsulfuration pathway. B6 is also required by approximately 140 other enzymes in human metabolism, including the GABA-synthesizing enzyme glutamate decarboxylase, the heme-synthesis enzyme ALA synthase, and the dopamine/serotonin-synthesizing aromatic amino acid decarboxylase. B6 deficiency therefore produces a pleiotropic clinical picture — elevated homocysteine, microcytic anemia, dermatitis, peripheral neuropathy, and seizures in severe deficiency.
Two ancillary cofactors deserve mention:
- Riboflavin (vitamin B2) — required as FAD by MTHFR itself. Suboptimal riboflavin status worsens the functional impact of MTHFR polymorphisms; riboflavin supplementation can partially compensate for the C677T variant by stabilizing the remaining enzyme.
- Betaine (trimethylglycine, TMG) — not a vitamin but a quaternary ammonium compound found in beets, wheat germ, spinach, and quinoa. Provides an alternative methyl source for the BHMT remethylation pathway, particularly important in liver and kidney. Cystic fibrosis patients, alcoholic liver disease patients, and patients with severe MTHFR deficiency may benefit from supplemental betaine (typical dose 500-3,000 mg/day, increasing to 6 g/day under medical supervision for homocystinuria).
For clinical work on adult patients with suspected methylation impairment, the standard laboratory panel is fasting homocysteine, MMA, serum B12, RBC folate, and serum B6 (PLP). Adding the MTHFR C677T and A1298C genotype is reasonable but not essential for treatment decisions — the homocysteine value is the bottom line.
DNA and Histone Methylation (Epigenetics)
The epigenome is the layer of inheritable chemical modifications imposed on DNA and its associated histone proteins that determines which genes are expressed in which cells at which times. Two of the four principal epigenetic marks — DNA cytosine methylation and histone lysine/arginine methylation — depend on SAMe as the methyl source. The other two (histone acetylation and chromatin remodeling) are SAMe-independent but are functionally coupled to the methylation marks.
DNA methylation is catalyzed by three principal DNA methyltransferases:
- DNMT1 — the maintenance methyltransferase that copies the methylation pattern of the parent strand onto the newly synthesized daughter strand during DNA replication. This is the mechanism by which methylation patterns are propagated across cell divisions, preserving cell-type identity.
- DNMT3A and DNMT3B — the de novo methyltransferases that establish new methylation patterns during embryogenesis, gametogenesis, and the limited de novo methylation that occurs in adult tissues. These are particularly important during the first weeks of pregnancy, when the global epigenetic landscape of the embryo is established.
The dependence on SAMe is absolute — both DNMT1 and the de novo enzymes are catalytically inactive without SAMe and are inhibited by accumulated SAH. The SAH/SAMe ratio (sometimes called the methylation potential) is therefore the operational driver of DNA methylation activity. In conditions of methionine deficiency or impaired remethylation, the SAH/SAMe ratio rises, DNMT activity falls, and DNA hypomethylation results. This has been documented in folate deficiency, B12 deficiency, severe MTHFR deficiency, alcoholic liver disease, and in dietary models of methionine restriction.
Histone methylation is more complex than DNA methylation because the marks can be either activating or repressive depending on which residue is methylated and how many methyl groups are added. H3K4me3 (histone 3, lysine 4, trimethylated) is an activating mark at transcription start sites. H3K27me3 is a repressive mark deposited by the Polycomb complex. H3K9me3 is a repressive mark associated with heterochromatin. All of these marks — both activating and repressive — require SAMe as the methyl source.
The clinical relevance of methylation-driven epigenetic regulation is most apparent during pregnancy. The neural tube closure event between days 21 and 28 post-conception requires extensive cellular proliferation and differentiation, which in turn requires extensive DNA methylation. The well-established protective effect of periconceptional folate supplementation against neural tube defects is mediated through the methionine cycle — adequate folate maintains adequate remethylation, adequate SAMe, and adequate DNA methylation capacity during this critical window. The dose-response relationship has been demonstrated in multiple large trials and is the basis for mandatory folate fortification of grain products in dozens of countries.
Beyond pregnancy, the epigenetic consequences of chronic methylation impairment in adults are a topic of ongoing research. Aberrant DNA methylation patterns have been documented in numerous cancers, cardiovascular disease, neurodegenerative disease, and accelerated aging (the "epigenetic clock" methods of Horvath and others). Whether dietary methionine, folate, B12, and B6 sufficiency can therapeutically modify these patterns at the adult-disease scale remains an active research area.
Neurotransmitter Synthesis via SAMe
Neurotransmitter biology is intimately tied to methylation. Three of the major monoamine systems — dopamine, norepinephrine/epinephrine, and serotonin/melatonin — have rate-limiting steps that are either directly SAMe-dependent or are tightly coupled to methionine-cycle metabolites:
- Epinephrine synthesis — the final step in catecholamine biosynthesis is the conversion of norepinephrine to epinephrine by phenylethanolamine N-methyltransferase (PNMT), which uses SAMe as the methyl donor. PNMT is strongly expressed in the adrenal medulla and in specific brainstem nuclei. SAMe limitation reduces epinephrine synthesis.
- Dopamine, norepinephrine, and epinephrine clearance — catechol-O-methyltransferase (COMT) inactivates these catecholamines by O-methylating the 3-hydroxyl of the catechol ring. COMT activity is genetically variable (the Val158Met polymorphism in COMT produces a 4-fold difference in enzyme activity between MM and VV homozygotes), and the COMT-Met variant (lower enzyme activity) is associated with higher prefrontal dopamine tone, anxiety phenotype, and certain pain sensitivity profiles. Impaired SAMe availability functionally mimics low-COMT genotype.
- Melatonin synthesis — the final step in the serotonin-to-melatonin pathway in the pineal gland is hydroxyindole-O-methyltransferase (HIOMT, also called ASMT), which uses SAMe to methylate the 5-hydroxyl of N-acetylserotonin. Reduced SAMe availability reduces evening melatonin synthesis.
- Serotonin precursor metabolism — tryptophan hydroxylase (TPH), the rate-limiting enzyme in serotonin synthesis, requires tetrahydrobiopterin (BH4) as cofactor. BH4 regeneration is folate-dependent and impaired in folate deficiency, indirectly linking serotonin synthesis to one-carbon metabolism. SAMe supplementation has been shown in animal and human studies to acutely raise CSF metabolites of serotonin, dopamine, and norepinephrine.
The integrated clinical consequence is that severe methylation impairment produces a recognizable neuropsychiatric phenotype — depression that is often refractory to SSRIs, fatigue, anxiety, attentional difficulties, and disrupted sleep with abnormal melatonin rhythm. This pattern is reported in some patients with severe MTHFR deficiency, in patients with chronic alcohol use (which depletes both SAMe and B-vitamin cofactors), and in patients with chronic gastrointestinal malabsorption affecting B12 absorption.
For the depression-specific clinical evidence on SAMe supplementation, see the dedicated section below on SAMe clinical trials and our Depression page.
Phosphatidylcholine and Membrane Synthesis
Every cellular and intracellular membrane in the human body is built primarily from phospholipids, and phosphatidylcholine (PC) is the most abundant phospholipid in essentially every cell type. There are two pathways by which the body produces PC:
- The CDP-choline pathway (Kennedy pathway) — uses dietary choline directly. Dietary choline is phosphorylated, then activated to CDP-choline, then conjugated with diacylglycerol to yield phosphatidylcholine. This pathway is the dominant PC source in most tissues.
- The PEMT pathway (phosphatidylethanolamine N-methyltransferase) — sequentially methylates phosphatidylethanolamine (PE) three times using SAMe as the methyl source to produce phosphatidylcholine. This pathway is most active in the liver, where it produces a structurally distinct PC molecular species (with more arachidonic acid in the sn-2 position) that is preferentially packaged into very-low-density lipoprotein (VLDL) particles for export.
The PEMT pathway has special significance in hepatic lipid metabolism. VLDL is the principal mechanism by which the liver exports triglycerides synthesized from dietary fat and de novo lipogenesis. VLDL assembly requires PC of the PEMT-derived molecular species. When the PEMT pathway is impaired — whether by SAMe deficiency, choline deficiency that forces the liver to rely on PEMT, or genetic PEMT polymorphisms — VLDL assembly stalls, triglycerides accumulate in the hepatocyte, and the histologic and metabolic picture of non-alcoholic fatty liver disease (NAFLD) develops.
This is the mechanistic basis for the methionine-choline-deficient (MCD) diet as the most-used animal model of NASH (non-alcoholic steatohepatitis). Removing both methionine (which feeds the PEMT pathway) and choline (which feeds the Kennedy pathway) from rodent feed produces histologic steatohepatitis with inflammation, ballooning, and fibrosis within four to eight weeks. The model is not perfect (the animals lose weight rather than gain it, the opposite of human NAFLD), but it demonstrates the centrality of methionine and choline to hepatic phospholipid and lipid metabolism.
For the full discussion of methionine and liver health, see our Liver Health deep-dive.
SAMe Clinical Trials (Depression, Liver, Osteoarthritis)
Pharmaceutical-grade SAMe (sold over the counter in the United States since 1999, prescription in Italy, Germany, and a number of other European countries) has been studied in three principal clinical applications:
- Major depression — meta-analyses pooling more than 40 placebo-controlled and active-comparator trials have consistently shown SAMe to be effective for major depressive disorder, with effect sizes in the range of standard tricyclic antidepressants. The Cochrane review and several systematic reviews place SAMe roughly equivalent to imipramine in efficacy with a substantially better tolerability profile. SAMe has also been studied as an SSRI augmenter, with the Papakostas 2010 American Journal of Psychiatry trial showing significant benefit for SSRI non-responders. Typical dosing is 400-1600 mg/day in divided doses (often started at 200 mg twice daily and titrated up to assess tolerability). The principal limitations are cost (SAMe is more expensive than generic SSRIs) and the requirement for stomach-friendly enteric-coated formulations because oral SAMe is acid-labile.
- Intrahepatic cholestasis — SAMe has been studied extensively in cholestatic liver disease, including intrahepatic cholestasis of pregnancy (ICP), primary biliary cholangitis, drug-induced cholestasis, and parenteral nutrition-induced cholestasis. The Mato 1999 Hepatology trial of SAMe in alcoholic cirrhosis showed improvement in survival and reduction in liver transplant need in Child-Pugh A and B patients. Subsequent trials in intrahepatic cholestasis of pregnancy show improvement in pruritus and biochemical markers, though ursodeoxycholic acid (UDCA) remains first-line. Mechanistically, SAMe supports hepatic phosphatidylcholine synthesis (membrane fluidity for canalicular bile excretion), supports glutathione synthesis, and provides methyl groups for bile acid conjugation.
- Osteoarthritis — meta-analyses including the Cochrane review have shown SAMe to be approximately as effective as NSAIDs (ibuprofen, naproxen, celecoxib) for symptom relief in knee and hip osteoarthritis, with a slower onset (4-6 weeks for full effect compared to days for NSAIDs) but a much more favorable gastrointestinal and cardiovascular safety profile. Typical OA dosing is 600-1200 mg/day. Mechanistically, SAMe contributes sulfur substrate to proteoglycan sulfation in cartilage and exerts anti-inflammatory effects through methylation-dependent gene regulation.
Across all three indications, the safety profile of SAMe in adult populations is excellent at standard therapeutic doses. The principal cautions are bipolar disorder (SAMe can precipitate or worsen mania, like any antidepressant), drug interactions with serotonergic medications (theoretical serotonin syndrome risk), and the practical issue of cost and the need for enteric-coated formulations.
Methylation Impairment: Clinical Pattern
Chronic methylation impairment produces a recognizable but non-specific clinical picture that affects multiple organ systems simultaneously. Patients may present with any combination of the following:
- Neuropsychiatric — depression often refractory to first-line antidepressants, anxiety, irritability, cognitive fog, attentional difficulties, fatigue, disrupted sleep architecture
- Cardiovascular — elevated fasting homocysteine, accelerated atherosclerosis, increased thrombotic risk, hypertension
- Hepatic — non-alcoholic fatty liver, elevated AST/ALT in the absence of viral hepatitis or alcohol, impaired drug clearance
- Reproductive — recurrent pregnancy loss, neural tube defects in offspring, male infertility (DNA methylation is essential for spermatogenesis)
- Histamine/hormone — histamine intolerance (impaired HNMT clearance, separate from DAO insufficiency), estrogen dominance symptoms (impaired COMT-mediated estrogen clearance)
- Energy and musculoskeletal — chronic fatigue, exercise intolerance, low muscle creatine, fibromyalgia-spectrum symptoms
- Hematologic — macrocytosis (elevated MCV) from impaired DNA synthesis, megaloblastic changes in severe B12 or folate deficiency
The work-up for suspected methylation impairment includes:
- Fasting plasma homocysteine (the single most useful test; normal <10 micromoles/L, optimal <7)
- Methylmalonic acid (MMA) to distinguish B12 deficiency from folate deficiency
- Serum vitamin B12 (interpret with awareness that normal-range B12 does not rule out functional deficiency — check MMA)
- RBC folate (better than serum folate for nutritional adequacy)
- Serum B6 (pyridoxal-5-phosphate) — an underused test
- Optionally, MTHFR C677T and A1298C genotyping
- Complete blood count with MCV (macrocytosis as functional marker)
- Liver enzymes and CMP
The therapeutic approach for confirmed methylation impairment combines repletion of any deficient cofactor, activated forms of folate and B12 (L-5-MTHF and methylcobalamin) to bypass MTHFR/MTR limitations, and consideration of SAMe supplementation directly for patients with neuropsychiatric symptoms. Betaine (trimethylglycine) is added for severe homocysteine elevation or hepatic indications. Follow-up homocysteine at 8-12 weeks confirms treatment response.
Practical SAMe and Methionine Supplementation
For most patients, optimizing methylation does not require methionine or SAMe supplementation as the first intervention. Optimizing dietary methionine through adequate protein intake (especially eggs, fish, poultry, and modest amounts of dairy or quality plant proteins) plus B-vitamin sufficiency through whole foods or a quality B-complex is usually sufficient. Direct SAMe supplementation is indicated when there is a specific clinical reason — depression, cholestatic liver disease, osteoarthritis, or documented severe methylation impairment with neuropsychiatric symptoms.
Practical considerations:
- Enteric coating is essential — SAMe is degraded by stomach acid. Quality SAMe products use enteric-coated tablets or capsules that release in the small intestine. Bargain SAMe products without enteric coating are largely wasted.
- Take on empty stomach — 30 minutes before breakfast or before lunch optimizes absorption.
- Start low, titrate up — begin at 200 mg once or twice daily, increase by 200 mg increments every 1-2 weeks based on tolerance and response. Most adults stabilize at 400-800 mg/day; depression dosing may go to 1600 mg/day.
- Pair with B-vitamin support — SAMe supplementation in the absence of adequate B12, folate, and B6 can paradoxically raise homocysteine by increasing the methylation flux without supporting the downstream remethylation or transsulfuration steps. Take a quality B-complex (with methylated folate and methyl-B12) alongside any SAMe regimen.
- Methionine supplementation directly is rarely needed when SAMe is the actual deficit. Methionine itself is the more appropriate intervention when there is dietary insufficiency (severe protein restriction, malabsorption, certain inherited disorders of methionine handling) but is not commonly used as a stand-alone supplement in adult practice.
Cautions and Drug Interactions
- Bipolar disorder — SAMe, like any antidepressant, can precipitate or worsen mania in patients with bipolar I or II disorder. Screen for personal or family history of mania before initiating; if bipolar is suspected, do not use without mood-stabilizer cover and specialist supervision.
- Serotonergic medication interactions — concurrent use of SAMe with SSRIs, SNRIs, MAOIs, tramadol, or triptans theoretically increases serotonin syndrome risk. The combined use of SAMe plus SSRI is common in clinical practice (the Papakostas 2010 augmentation trial established efficacy and safety in this setting) but should be monitored for emergent agitation, hyperreflexia, or autonomic instability.
- Levodopa — SAMe can theoretically reduce levodopa effectiveness by increasing peripheral COMT-mediated levodopa methylation, reducing the fraction reaching the CNS. Patients with Parkinson's on levodopa should use SAMe only under specialist guidance.
- Pregnancy and lactation — SAMe is endogenous and standard prenatal vitamin preparations contain folate and B12 specifically to support methylation in pregnancy. SAMe itself has been used for intrahepatic cholestasis of pregnancy with apparent safety, but routine SAMe supplementation in pregnancy is not standard and should be specialist-directed.
- Methionine and homocysteine — high-dose methionine (much higher than dietary intake) acutely raises homocysteine, which is the basis for the "methionine loading test" once used to diagnose CBS deficiency. This is generally not a concern at supplemental doses but is part of why direct methionine supplementation is less commonly used than SAMe.
- Methionine restriction — there is an active research literature on dietary methionine restriction as a longevity intervention in mice and rats, with the relevant translational question being whether high methionine intake (typical of high-protein omnivorous diets) is itself a risk factor for accelerated aging. This nuance is addressed in our Benefits Deep Dive hub page in the methionine-restriction paradox section.
Key Research Papers
- Cantoni GL (1953). S-adenosylmethionine; a new intermediate formed enzymatically from L-methionine and adenosinetriphosphate. Journal of Biological Chemistry. — PubMed
- Mato JM et al. (1999). S-adenosylmethionine in alcoholic liver cirrhosis: a randomized, placebo-controlled, double-blind, multicenter clinical trial. Journal of Hepatology. — PubMed
- Papakostas GI et al. (2010). S-adenosyl methionine (SAMe) augmentation of serotonin reuptake inhibitors for antidepressant nonresponders with major depressive disorder. American Journal of Psychiatry. — PubMed
- Frosst P et al. (1995). A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nature Genetics. — PubMed
- Selhub J (1999). Homocysteine metabolism. Annual Review of Nutrition. — PubMed
- Lu SC, Mato JM (2012). S-adenosylmethionine in liver health, injury, and cancer. Physiological Reviews. — PubMed
- Sharma A et al. (2017). S-Adenosylmethionine (SAMe) for neuropsychiatric disorders: a clinician-oriented review of research. Journal of Clinical Psychiatry. — PubMed
- Galizia I et al. (2016). S-adenosyl methionine (SAMe) for depression in adults. Cochrane Database of Systematic Reviews. — PubMed
- Soeken KL et al. (2002). Safety and efficacy of S-adenosylmethionine (SAMe) for osteoarthritis. Journal of Family Practice. — PubMed
- Mato JM, Martínez-Chantar ML, Lu SC (2008). Methionine metabolism and liver disease. Annual Review of Nutrition. — PubMed
- Stipanuk MH (2004). Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annual Review of Nutrition. — PubMed
- Friso S et al. (2002). A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. PNAS. — PubMed
- Reik W (2007). Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. — PubMed
PubMed Topic Searches
- PubMed: SAMe universal methyl donor
- PubMed: Methionine cycle remethylation/transsulfuration
- PubMed: MTHFR polymorphisms and homocysteine
- PubMed: DNA methylation and SAMe
- PubMed: PEMT phosphatidylcholine and liver
- PubMed: SAMe for depression trials
Connections
- Methionine Overview
- Methionine Benefits Hub
- Methionine for Liver Health
- Methionine for Detoxification
- Methionine for Hair and Nails
- Cysteine
- Glycine
- Taurine
- Vitamin B6
- Vitamin B12
- Homocysteine Lab Test
- NAC & Glutathione
- Depression
- Sulfur
- Creatine
- All Amino Acids