Tuberous Sclerosis Complex
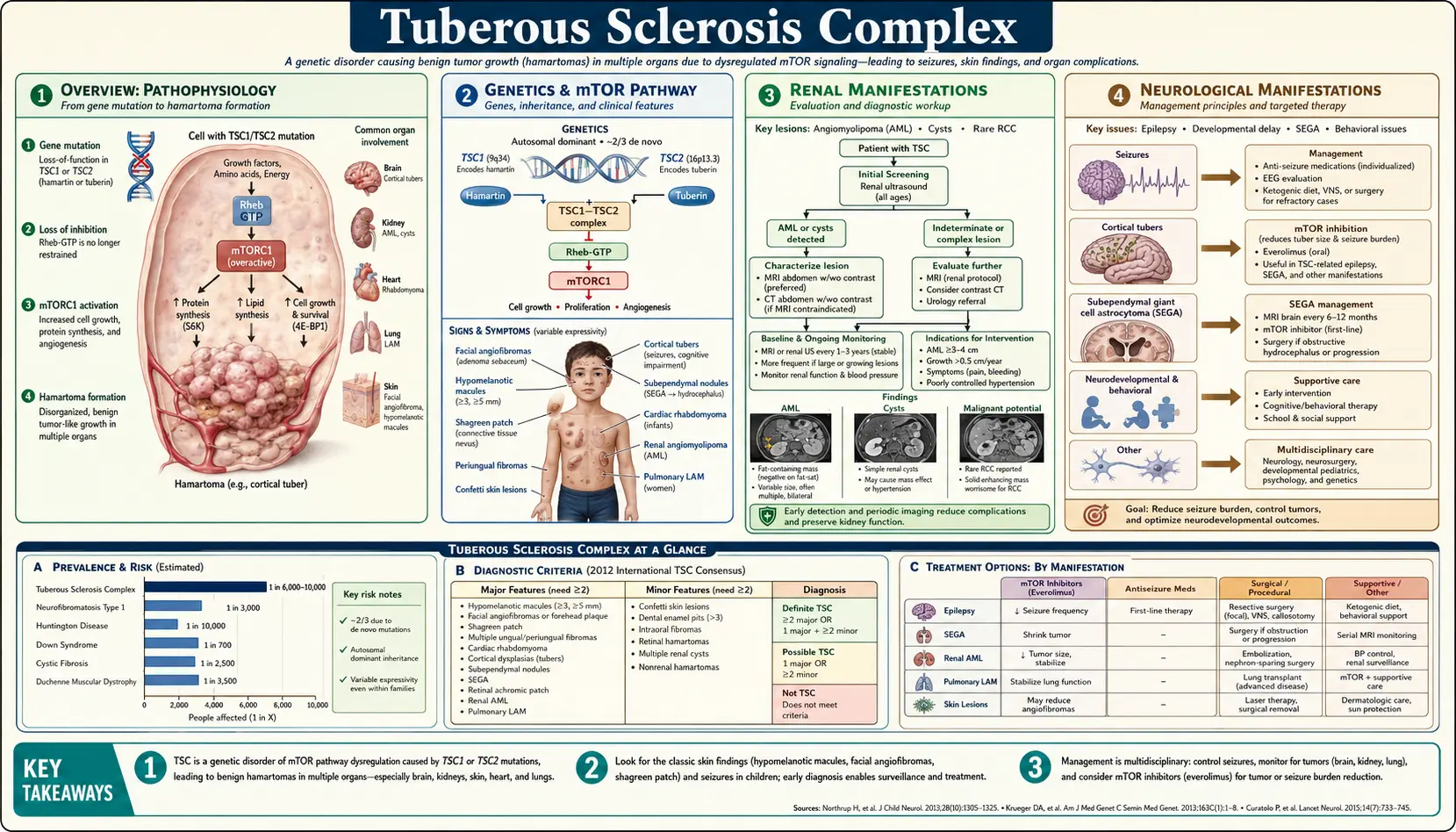
Tuberous sclerosis complex (TSC) is an autosomal dominant multisystem disorder caused by loss-of-function mutations in TSC1 or TSC2 genes, resulting in dysregulated mTORC1 signaling and benign hamartoma formation in the brain, kidneys, lungs, skin, and heart. The disorder affects approximately 1 in 6,000 individuals worldwide and is a leading genetic cause of epilepsy, intellectual disability, and renal angiomyolipomas.
Table of Contents
- Overview
- Genetics and mTOR Pathway
- Renal Manifestations
- Neurological Manifestations
- Dermatological Manifestations
- Pulmonary Manifestations (LAM)
- Cardiac and Other Manifestations
- Diagnosis
- Treatment
- Surveillance and Monitoring
- Research Papers (PubMed searches)
- References
- Featured Videos
1. Overview
Tuberous sclerosis complex (TSC, OMIM #191100/#613254) is a rare autosomal dominant neurocutaneous syndrome characterized by formation of benign hamartomas in multiple organs. Named for the “tuber-like” calcified cortical lesions observed on pathology (first described by Désiré-Magloire Bourneville in 1880), TSC has a population prevalence of ~1 in 6,000 and affects approximately 1 million people worldwide. New mutations account for two-thirds of cases (de novo). The disorder spans a wide clinical spectrum — from minimally symptomatic to severely affected — and is the second most common genetic cause of epilepsy after Dravet syndrome. The molecular basis (TSC1/TSC2 mutations → mTORC1 hyperactivation) has enabled targeted therapy with mTOR inhibitors (everolimus, sirolimus), transforming management.
2. Genetics and mTOR Pathway
Two causative genes: TSC1 (chromosome 9q34, encodes hamartin) and TSC2 (chromosome 16p13.3, encodes tuberin). TSC2 mutations are ~4× more common than TSC1 and correlate with more severe phenotype (lower IQ, more renal AMLs, earlier ESRD, more retinal hamartomas). De novo mutations account for ~65–70% of all TSC cases (neither parent affected). Approximately 10–15% of patients have no identifiable mutation due to somatic mosaicism, deep intronic variants, or structural rearrangements.
Molecular mechanism: Hamartin and tuberin form a functional dimer (TSC1–TSC2 complex) that acts as a GTPase-activating protein (GAP) for Rheb (Ras homolog enriched in brain). Active Rheb-GTP activates mTORC1 (mechanistic target of rapamycin complex 1), a master regulator of protein synthesis, cell growth, autophagy, and metabolism. Loss of TSC1 or TSC2 function eliminates the GAP activity → constitutively active Rheb → chronic mTORC1 hyperactivation → uncontrolled cell growth, proliferation, and survival → hamartoma formation. Second-hit somatic mutations in the remaining wild-type allele occur in hamartoma cells (loss of heterozygosity), explaining the focal nature of lesions despite a germline mutation.
3. Renal Manifestations
Renal disease is a leading cause of TSC-related mortality, affecting ~1% of TSC patients annually from life-threatening complications.
Angiomyolipomas (AMLs): Found in 55–80% of TSC patients. Benign mesenchymal tumors composed of abnormal blood vessels (tortuous, thin-walled, aneurysm-prone), smooth muscle cells, and adipose tissue. Typically bilateral and multiple. Enlarge over a lifetime at a linear growth rate of ~0.8 cm/year. Critical size threshold: AMLs ≥4 cm carry high risk of spontaneous hemorrhage (Wunderlich syndrome — retroperitoneal bleeding, potentially life-threatening). Risk factors for bleeding: size ≥4 cm, aneurysm ≥0.5 cm. Epithelioid AML variant is rare, potentially malignant, and may metastasize.
Renal cysts: Present in 20–30% of TSC patients. Typically multiple bilateral simple cysts. May be associated with a TSC2/PKD1 contiguous gene deletion syndrome (chromosome 16p13.3 deletion spanning both genes) — causes severe early-onset polycystic kidney disease mimicking ARPKD, leading to renal failure in childhood. A PKD1-adjacent TSC2 deletion must be suspected when TSC is associated with severe childhood polycystic disease.
Renal cell carcinoma (RCC): Risk 2–4× that of the general population. May occur at a younger age and be bilateral or multifocal. Most are clear cell or chromophobe type. Annual screening is required.
Renal failure: Occurs in ~1–2% of TSC patients from AML replacement of renal parenchyma, cystic disease (TSC2/PKD1), or bilateral AML hemorrhage.
4. Neurological Manifestations
Brain involvement is the primary driver of morbidity in TSC.
Cortical tubers: Focal dysplastic cortical lesions (disorganized neurons and giant cells) resulting from impaired neuronal migration and maturation. Found in 80–90% of TSC patients. Number and location correlate with epilepsy severity and cognitive outcomes. MRI shows T2-hyperintense cortical/subcortical lesions that are pathognomonic.
Subependymal nodules (SENs): Hamartomatous nodules along the ventricular walls that calcify with age (“candle drippings” on CT). Found in 80% of TSC patients. Usually asymptomatic but can evolve into SEGAs.
Subependymal giant cell astrocytomas (SEGAs): Low-grade tumors arising from SENs at the foramen of Monro. Occur in 5–20% of TSC patients. Growth can cause obstructive hydrocephalus and raised intracranial pressure — a medical emergency. Treatment: everolimus (first-line for growing SEGAs) or surgical resection.
Epilepsy: Affects 90% of TSC patients; onset in infancy in >60% (infantile spasms with hypsarrhythmia on EEG in the first year of life in 30–35%). Infantile spasms are treated urgently with vigabatrin (first-line for TSC-associated infantile spasms, per UKISS trial and TSC Consensus guidelines) ± ACTH. Drug-resistant focal epilepsy from tubers is common. Epilepsy surgery (tuber resection) and dietary therapies (ketogenic diet) benefit selected patients. Everolimus demonstrated efficacy for refractory seizures in the EXIST-3 trial.
Neurodevelopmental outcomes: Intellectual disability in ~40–50% (IQ ranges widely). Autism spectrum disorder in 40–50% — a higher rate than any other single-gene cause of autism. ADHD, anxiety, and sleep disorders are common. The TSC-Associated Neuropsychiatric Disorders (TAND) framework guides structured assessment.
5. Dermatological Manifestations
Skin findings are often the first clinical sign leading to a TSC diagnosis.
Hypomelanotic macules (“ash-leaf spots”): Oval or lance-ovate hypopigmented patches (3–15 mm), present at birth in >90% of TSC patients. Best visualized under Wood’s lamp (UV light). Three or more hypomelanotic macules constitute a major diagnostic criterion.
Facial angiofibromas: Reddish-brown papules distributed symmetrically across the nose, cheeks, and nasolabial folds (“butterfly rash”). Present in 75% of patients, typically appearing in childhood (ages 2–5). Composed of fibrous tissue and ectatic blood vessels — not acne. Can cause significant cosmetic distress. Topical rapamycin (sirolimus) cream reduces angiofibromas (RIST trial and multiple RCTs).
Shagreen patches: Connective tissue nevi — irregularly surfaced, skin-colored plaques on the lower back or lumbosacral region with an “orange peel” or “leather” texture. Present in 20–30% of patients.
Ungual (periungual) fibromas (Koenen tumors): Flesh-colored fibrous outgrowths from the nail beds of fingers or toes. Develop in adolescence or adulthood, found in 15–20% of patients. Pathognomonic when present.
Confetti lesions: Multiple small (1–3 mm) hypopigmented macules on the extremities; present in ~10% of patients.
Fibrous cephalic plaques: Irregular, raised, skin-colored to yellowish plaques on the forehead or scalp.
6. Pulmonary Manifestations (LAM)
Lymphangioleiomyomatosis (LAM): Proliferation of abnormal TSC2-mutant smooth muscle-like LAM cells throughout the lung parenchyma. LAM occurs virtually exclusively in females, affecting ~30–40% of adult TSC women by age 40. It is characterized by progressive bilateral cystic lung destruction and presents with progressive dyspnea on exertion, recurrent pneumothorax (hallmark — occurring in 50–70% of symptomatic LAM patients), and chylous pleural effusions.
Imaging: High-resolution CT (HRCT) shows multiple bilateral thin-walled cysts throughout the lungs, distinguishable from emphysema by their round, uniform shape and normal intervening parenchyma. Lung biopsy is not usually required if clinical and radiologic criteria are met and serum VEGF-D is ≥800 pg/mL or HMB45-positive cells are found in bronchoalveolar lavage.
Treatment: Sirolimus slows the decline in FEV1 and improves quality of life (MILES trial, McCormack et al., N Engl J Med 2011). Lung transplantation is reserved for advanced disease (LAM may recur in the donor lung). Exogenous estrogen should be avoided as it accelerates progression.
TSC-LAM vs. sporadic LAM: Sporadic LAM (no TSC germline) arises from somatic biallelic TSC2 loss in LAM cells; it tends to present later and may be more severe.
7. Cardiac and Other Manifestations
Cardiac rhabdomyomas: Benign intracardiac hamartomas found in 50–65% of newborns with TSC, often diagnosed prenatally on fetal echocardiography. Multiple, usually located in ventricular walls. Most regress spontaneously by childhood — the majority are asymptomatic by age 6. Fetal arrhythmia or hydrops may occur in rare cases with massive tumors. Wolff-Parkinson-White (WPW) pattern or other arrhythmias may occur if tumors involve the conduction system. Prenatal diagnosis of multiple cardiac rhabdomyomas should prompt TSC genetic testing.
Hepatic angiomyolipomas: Similar to renal AMLs; present in ~15% of TSC patients. Usually asymptomatic and rarely require intervention.
Bone cysts and sclerotic lesions: Incidental finding on imaging in >50% of patients; rarely symptomatic.
Retinal astrocytic hamartomas: Present in 30–50% of TSC patients. Usually asymptomatic and rarely affect vision. Mulberry-like calcified nodules near the optic disc, or flat semitransparent lesions. Calcified forms are pathognomonic.
Aortic aneurysm: Rare association, particularly in TSC2 patients.
8. Diagnosis
Clinical diagnostic criteria are defined by the Northrup and Krueger 2012 International TSC Consensus: Definite TSC = 2 major features OR 1 major + 2 minor features.
Major features (11): hypomelanotic macules ≥3 (≥5 mm); facial angiofibromas or fibrous cephalic plaque; ungual fibromas ≥2; shagreen patch; multiple retinal hamartomas; cortical dysplasia (tuber or radial migration lines); subependymal nodule; subependymal giant cell astrocytoma; cardiac rhabdomyoma; lymphangioleiomyomatosis; angiomyolipomas ≥2.
Minor features (6): confetti skin lesions; dental enamel pits ≥3; intraoral fibromas ≥2; retinal achromic patch; multiple renal cysts; nonrenal hamartomas.
Genetic diagnosis: A pathogenic TSC1 or TSC2 variant alone is sufficient for definitive diagnosis regardless of clinical features.
Differential diagnosis: Neurofibromatosis type 1 (café-au-lait spots, not ash-leaf; Lisch nodules; no renal AMLs), Sturge-Weber syndrome, isolated sporadic LAM, isolated renal AML.
Neuroimaging: Brain MRI (cortical tubers, SENs, SEGAs). Abdominal MRI (preferred over CT for AML and renal cyst assessment — avoids radiation, better fat characterization). Chest HRCT (LAM screening). Echocardiography (newborns). Ophthalmology examination. EEG. Neuropsychological assessment.
9. Treatment
mTOR Inhibitors (cornerstone of targeted therapy)
Everolimus (Afinitor): Oral mTOR inhibitor. FDA-approved for: (1) growing SEGA — EXIST-1 trial: 35% volume reduction vs. 0% placebo; (2) renal AMLs ≥3 cm not requiring urgent surgery — EXIST-2 trial: 42% volume reduction; (3) refractory focal seizures — EXIST-3 trial: ≥50% seizure reduction in 40% of patients vs. 15% placebo. Dose: 4.5 mg/m² (SEGA), 10 mg/day (AML/LAM). Side effects: aphthous stomatitis, infections, hyperlipidemia, pulmonary toxicity, impaired wound healing. Tumor regrowth occurs after stopping — long-term or continuous therapy is often required.
Sirolimus (rapamycin): Same mechanism as everolimus. FDA-approved for LAM (MILES trial — stabilizes FEV1 decline). Topical sirolimus gel for facial angiofibromas (RIST and other trials). Used off-label for renal AMLs.
AML Management
AMLs <3 cm: surveillance with annual MRI. AMLs ≥3 cm or growing: first-line everolimus (reduces need for invasive procedures). Acute hemorrhage (Wunderlich syndrome): selective arterial embolization (SAE) — embolize the feeding artery; nephron-sparing; surgery (partial nephrectomy) if embolization fails or tumor is epithelioid. Nephrectomy is avoided when possible to preserve renal function. Prophylactic embolization for AMLs ≥4 cm with aneurysm is debated; everolimus is preferred over prophylactic embolization in current guidelines.
Epilepsy
Vigabatrin is first-line for infantile spasms in TSC (UKISS trial: 95% spasm cessation with vigabatrin vs. 73% with hormonal therapy). Standard antiseizure medications (carbamazepine, levetiracetam, lamotrigine) for ongoing seizures. Everolimus for refractory seizures. Epilepsy surgery for drug-resistant focal seizures localized to a resectable tuber. Ketogenic diet and vagus nerve stimulation are additional options.
SEGA
Everolimus is first-line for asymptomatic growing SEGA. Surgical resection is indicated for symptomatic SEGA causing acute hydrocephalus.
LAM
Sirolimus for progressive LAM (FEV1 <70% predicted or rapid decline). Supplemental oxygen as needed. Pleurodesis for recurrent pneumothorax. Lung transplantation for advanced disease.
10. Surveillance and Monitoring
Based on International TSC Consensus (2012) recommendations:
Renal: MRI abdomen every 1–3 years (AML and cyst monitoring). Annual blood pressure and renal function tests. More frequent surveillance if AMLs are ≥3 cm, bilateral, or actively growing.
Brain: MRI brain every 1–3 years in children (more frequent if SEGA is present or growing). Annual EEG if seizure control is inadequate. Neuropsychological assessment every 3 years.
Pulmonary (females ≥18 years): Baseline HRCT chest; if cysts are present, spirometry annually and HRCT every 2–3 years; measure serum VEGF-D.
Cardiac: Echocardiogram in neonates and infants; ECG if arrhythmia symptoms develop. After rhabdomyoma regression is confirmed, cardiac surveillance can be discontinued unless arrhythmia is present.
Dermatology: Annual skin examination. Ophthalmology: Baseline and periodic retinal examination. Dentistry: Annual dental examination (enamel pits, gingival fibromas).
Neurodevelopmental (TAND): Formal assessment at key developmental stages (infancy, pre-school, school entry, adolescence, adulthood). The TAND Checklist guides primary care screening.
11. Research Papers (PubMed searches)
- tuberous sclerosis complex TSC1 TSC2 mTOR
- tuberous sclerosis renal angiomyolipoma everolimus EXIST-2
- tuberous sclerosis epilepsy vigabatrin infantile spasms
- TSC lymphangioleiomyomatosis sirolimus MILES trial
- subependymal giant cell astrocytoma TSC everolimus EXIST-1
- tuberous sclerosis autism neuropsychiatric
12. References
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):243–254. PMID: 24053982. https://doi.org/10.1016/j.pediatrneurol.2013.08.001
- Franz DN, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381(9861):125–132. PMID: 23158522. https://doi.org/10.1016/S0140-6736(12)61134-9
- Bissler JJ, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817–824. PMID: 23312829. https://doi.org/10.1016/S0140-6736(12)61767-X
- French JA, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016;388(10056):2153–2163. PMID: 27613521. https://doi.org/10.1016/S0140-6736(16)31419-2
- McCormack FX, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis (MILES Trial). N Engl J Med. 2011;364(17):1595–1606. PMID: 21410393. https://doi.org/10.1056/NEJMoa1100391
- O’Callaghan FJK, et al. The effect of lead time to treatment and treatment duration response for infantile spasms caused by tuberous sclerosis (UKISS trial). Epilepsia. 2011;52(8):1359–1365. PMID: 21682737. https://doi.org/10.1111/j.1528-1167.2011.03136.x
- Krueger DA, et al. Everolimus for treatment of epilepsy associated with tuberous sclerosis complex (EXIST-3): final results. Neurology. 2020;95(13):e1755–e1767. PMID: 32868465. https://doi.org/10.1212/WNL.0000000000010595
- Henske EP, et al. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035. PMID: 27226234. https://doi.org/10.1038/nrdp.2016.35
- Roach ES, et al. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998;13(12):624–628. PMID: 9881533. https://doi.org/10.1177/088307389801301206
- Kingswood JC, et al. Renal findings in a European tuberous sclerosis complex (TSC) registry. Nephrol Dial Transplant. 2019;34(3):502–508. PMID: 29409041. https://doi.org/10.1093/ndt/gfx336
- Dabora SL, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68(1):64–80. PMID: 11112665. https://doi.org/10.1086/316951
- Bissler JJ, Kingswood JC. Renal angiomyolipomata. Kidney Int. 2004;66(3):924–934. PMID: 15327392. https://doi.org/10.1111/j.1523-1755.2004.00838.x
Connections
- Nephrology & Hepatology
- Chronic Kidney Disease
- IgA Nephropathy
- Alport Syndrome
- Fabry Disease
- Epilepsy
- Lymphangioleiomyomatosis (LAM)
- Renal Cell Carcinoma
- Kidney Function Tests