Fabry Disease
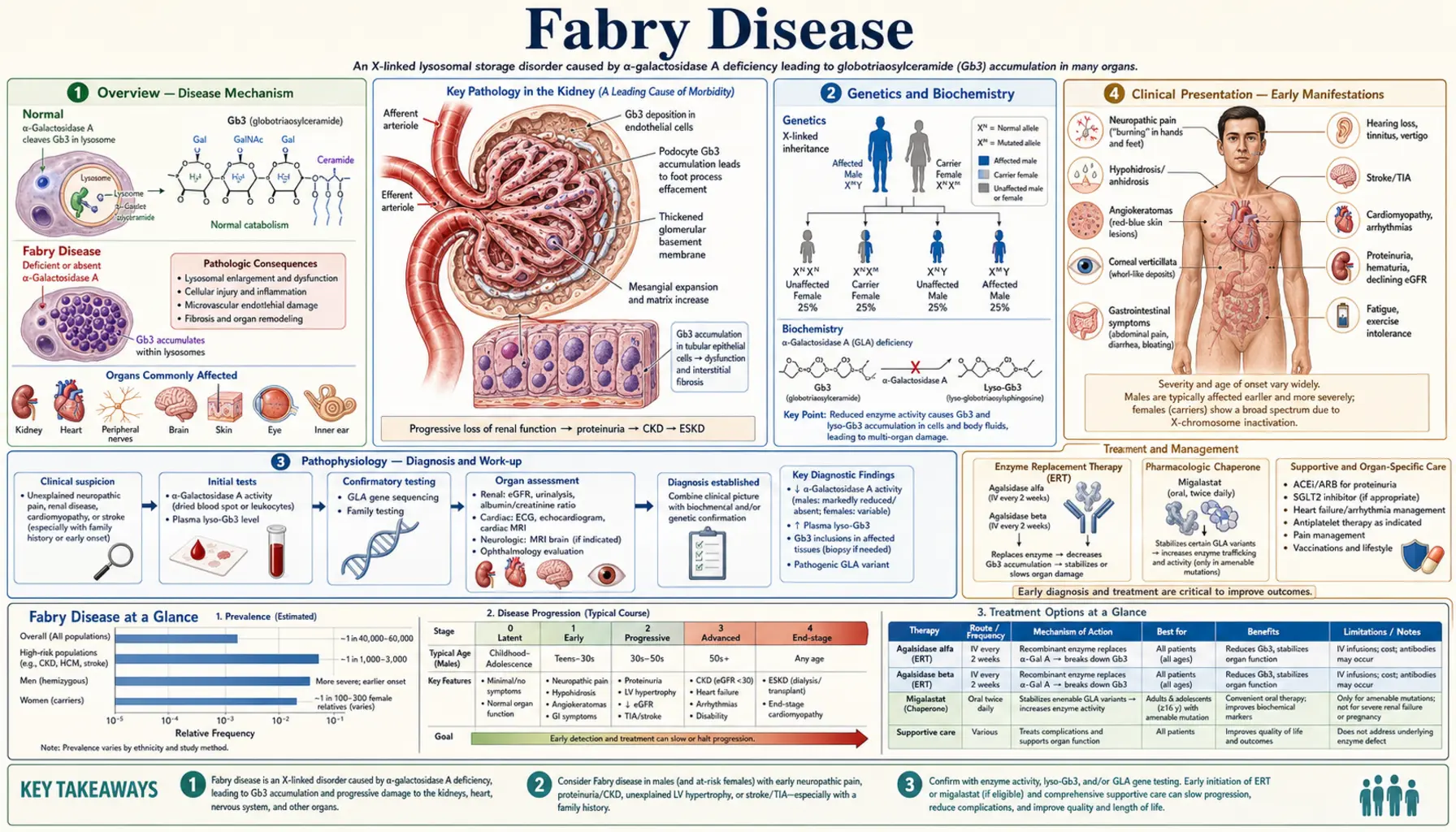
Fabry disease is an X-linked lysosomal storage disorder caused by deficiency of alpha-galactosidase A (alpha-Gal A, encoded by the GLA gene), leading to progressive intracellular accumulation of globotriaosylceramide (Gb3/GL-3) and its deacylated form lyso-Gb3 in endothelial, smooth muscle, cardiac, and renal cells. The disorder causes a multisystem vasculopathy affecting the kidneys, heart, nervous system, and skin, with untreated males typically developing end-stage renal disease and premature cardiovascular death.
Table of Contents
- Overview
- Genetics and Biochemistry
- Pathophysiology
- Clinical Presentation — Early Manifestations
- Organ Involvement — Kidney
- Organ Involvement — Heart
- Organ Involvement — Nervous System and Stroke
- Diagnosis
- Treatment
- Monitoring and Follow-Up
- Research Papers (PubMed searches)
- References
- Featured Videos
1. Overview
Fabry disease (Anderson-Fabry disease, OMIM #301500) is a rare X-linked lysosomal storage disorder with an estimated incidence of 1 in 40,000–120,000 male births in classic form; newborn screening studies suggest a broader prevalence of 1 in 1,500–3,100 when atypical/late-onset variants are included. Described independently by Johannes Fabry (Germany) and William Anderson (UK) in 1898. The disorder is caused by mutations in the GLA gene (chromosome Xq22.1), producing deficient or absent alpha-galactosidase A (alpha-Gal A) enzyme activity. This results in lysosomal accumulation of globotriaosylceramide (Gb3, also called GL-3) throughout the body, particularly in vascular endothelial and smooth muscle cells, podocytes, cardiomyocytes, and neurons. Classic Fabry disease in hemizygous males causes neuropathic pain, angiokeratomas, cornea verticillata, progressive kidney disease (ESRD by 40s in untreated males), hypertrophic cardiomyopathy, and early stroke. Enzyme replacement therapy and the oral chaperone migalastat have transformed the prognosis for treatable patients.
2. Genetics and Biochemistry
X-linked inheritance: GLA gene on chromosome Xq22.1; >1,000 pathogenic variants identified (missense, nonsense, splicing, insertions/deletions). Hemizygous males (one X chromosome): classic or atypical disease depending on residual enzyme activity. Heterozygous females: phenotype ranges from asymptomatic carrier to severe classic disease (X-inactivation determines severity). Neither parent-of-origin effect applies uniformly.
Classic vs. atypical variants:
- Classic Fabry (males): alpha-Gal A activity <1% of normal. Full multisystem involvement. Childhood/adolescent onset of acroparesthesias, angiokeratomas.
- Atypical/late-onset variants (commonly p.A143T, p.R112H, IVS4+919 G→A): residual alpha-Gal A activity 1–15%. Predominantly cardiac or renal involvement (late-onset cardiomyopathy or nephropathy); absent or minimal early neuropathic symptoms. Particularly prevalent in certain populations (p.A143T in Black males — uncertain pathogenicity; IVS4+919 common in Taiwan).
Biochemistry: Alpha-Gal A is a lysosomal hydrolase that cleaves terminal galactose residues from glycosphingolipids, primarily converting Gb3 (globotriaosylceramide, ceramide-glucose-galactose-galactose) to lactosylceramide. Deficiency causes Gb3 to accumulate within lysosomes. Gb3 accumulation in endothelial and smooth muscle cells causes vasculopathy; in podocytes causes progressive nephropathy; in cardiomyocytes causes hypertrophic cardiomyopathy; in neurons causes neuropathic pain. Lyso-Gb3 (deacylated form) is a more sensitive biomarker (elevated in males and female carriers) and promotes additional cellular injury pathways.
3. Pathophysiology
Gb3 accumulates within secondary lysosomes in virtually all cell types. The predominant pathological consequences:
Vascular endothelium and smooth muscle: Gb3-laden endothelial cells swell and occlude small vessel lumens (microangiopathy). This drives ischemia in the kidney (tubular atrophy, glomerulosclerosis), heart (myocardial ischemia despite normal coronaries), and brain (lacunar infarcts, white matter lesions, posterior circulation strokes). Vasculopathy is progressive and underlies the multiorgan failure.
Podocyte injury: Podocytes are among the most heavily Gb3-loaded cells in the kidney. Gb3 disrupts lysosomal function, induces autophagy failure, and triggers podocyte apoptosis → podocyte depletion → focal segmental glomerulosclerosis (FSGS) pattern → proteinuria → progressive CKD.
Cardiomyocytes: Gb3 accumulation causes myocyte hypertrophy and fibrosis → concentric left ventricular hypertrophy (LVH). Gb3 in the conduction system → arrhythmias (AV block, atrial fibrillation, Wolff-Parkinson-White-like patterns).
Dorsal root ganglia neurons: Gb3 accumulation → small fiber neuropathy → burning neuropathic pain (acroparesthesias), autonomic dysfunction (hypohidrosis, gastroparesis, orthostatic hypotension).
Lyso-Gb3: More water-soluble than Gb3; more readily detectable in plasma and urine. Acts as a signaling molecule that activates smooth muscle cell proliferation, inhibits K+ channels in sensory neurons (contributing to pain), and promotes Gb3 synthesis — amplifying disease.
4. Clinical Presentation — Early Manifestations
Classic Fabry disease in males presents in childhood and adolescence with:
Neuropathic pain (acroparesthesias): Burning, lancinating pain in hands and feet, characteristically worse with heat, fever, exercise, or emotional stress. Episodic “Fabry crises” (severe pain attacks lasting hours to days, precipitated by fever or exercise). Cause: small fiber neuropathy from Gb3 in dorsal root ganglia. Often the first and most disabling symptom; may begin as early as age 3–7. Frequently misdiagnosed as arthritis, rheumatic fever, or growing pains.
Hypohidrosis (reduced sweating): Due to Gb3 in eccrine sweat glands and autonomic ganglia. Causes heat intolerance — patients cannot regulate temperature during exercise or hot weather. Heat intolerance combined with acroparesthesias in a young male is a classic Fabry presentation.
Angiokeratomas: Small dark red to blue-black telangiectatic papules that do not blanch on pressure. Distributed classically in the “bathing trunk” area — lower abdomen, buttocks, genitalia, and upper thighs — and on lips/oral mucosa. Present in 70% of classic males, developing in teens. Pathognomonic when combined with acroparesthesias.
Cornea verticillata (corneal whorl): Whorled pattern of cream-colored corneal epithelial deposits (best seen on slit-lamp). Found in >90% of hemizygous males and >70% of carrier females. Asymptomatic. Also caused by amiodarone, chloroquine — must be distinguished.
Gastrointestinal: Nausea, vomiting, diarrhea, early satiety (Gb3 in autonomic GI ganglia). Often dismissed as irritable bowel. Common in childhood and adolescence.
5. Organ Involvement — Kidney
Renal disease is the second leading cause of death in untreated Fabry males (after cardiac disease).
Progression: Microalbuminuria typically appears in the late teens/20s in males. Proteinuria increases through 20s–30s. CKD progresses: eGFR decline rate ~3–5 mL/min/1.73m²/year in untreated males. ESRD in untreated males: median age 38–42 years. In treated males: ERT slows decline but does not stop it; stabilization best in those with eGFR >60 at treatment start and minimal proteinuria.
Histopathology: Kidney biopsy pathognomonic — electron microscopy shows zebra bodies (lamellar, concentric myelin-like inclusions within lysosomes of podocytes, mesangial cells, endothelial cells, tubular cells). Light microscopy: podocyte cytoplasm appears foamy/vacuolated on H&E. PAS stain shows glycolipid-laden cells. Immunofluorescence: Gb3 staining positive. Pattern: focal segmental glomerulosclerosis (FSGS) appearance + tubular atrophy as disease advances.
Proteinuria as key prognostic marker: Proteinuria >1 g/day strongly predicts faster progression to ESRD. RAAS blockade reduces proteinuria and slows CKD in Fabry as in other proteinuric nephropathies.
Dialysis and transplant: Renal replacement therapy outcomes are good in Fabry patients. Kidney transplant — allograft is free of Fabry disease (no GLA mutation). However, extrarenal manifestations (cardiac, neurological) continue to progress post-transplant — ERT should be continued. Recurrence in allograft does not occur, unlike most hereditary GN.
6. Organ Involvement — Heart
Cardiac disease is the leading cause of death in Fabry patients (responsible for ~40–50% of deaths).
Hypertrophic cardiomyopathy (HCM): Concentric LVH develops in the 3rd–4th decade in males (later in females). LV wall thickness increases progressively. Clinically: exertional dyspnea, chest pain, near-syncope. Echo: increased interventricular septal thickness and posterior wall thickness; preserved or reduced systolic function with impaired diastolic function in advanced disease. CMR: late gadolinium enhancement (LGE) in infero-lateral mid-myocardium (“Fabry pattern”) distinguishes from sarcomeric HCM. LGE correlates with fibrosis and predicts arrhythmia risk.
Arrhythmias: Bradyarrhythmias (sinus bradycardia, AV block from Gb3 in conduction system); tachyarrhythmias (atrial fibrillation in 10–20% of Fabry adults; ventricular tachycardia/fibrillation — rare but risk of sudden death). EKG: shortened PR interval (pseudo-WPW pattern), LVH voltage criteria, ST-T changes.
Heart failure: Late complication. Mixed systolic + diastolic dysfunction. Patients may require implantable cardiac defibrillator (ICD) for sudden death prevention.
Important clinical pearl: Fabry disease may cause HCM phenotype in 0.5–1% of patients presenting with “unexplained HCM” — Fabry should be screened in all unexplained HCM (especially males with reduced alpha-Gal A activity on dried blood spot).
7. Organ Involvement — Nervous System and Stroke
Cerebrovascular disease: Stroke and TIA occur in Fabry patients at a mean age of 33–46 years in males — far younger than the general population. Mechanism: small vessel vasculopathy (cerebral microangiopathy) + cardiac embolism (AF, LV thrombus). Pattern: posterior circulation strokes (basilar artery territory, posterior cerebral artery) are disproportionately common in Fabry. White matter lesions (leukoencephalopathy) on MRI T2/FLAIR — dolichoectasia of basilar artery is a characteristic finding. Fabry disease should be screened in cryptogenic stroke in young adults (age <50).
Small fiber neuropathy: Reduced intraepidermal nerve fiber density on skin biopsy. Autonomic neuropathy: reduced heart rate variability, orthostatic hypotension, hypohidrosis, gastroparesis, neurogenic bladder.
CNS: Cognitive impairment, depression, anxiety are common but often under-recognized. May relate to chronic pain, white matter disease, and/or direct Gb3 effects on CNS.
Tinnitus and hearing loss: Sensorineural hearing loss and tinnitus occur in ~50% of Fabry patients; usually mild-to-moderate.
8. Diagnosis
Diagnosis requires a high index of suspicion given the rarity and multisystem nature.
Alpha-galactosidase A enzyme activity:
- Hemizygous males: alpha-Gal A activity in leukocytes (WBC) or dried blood spot (DBS) — markedly low or absent in classic disease; may be normal in females and some atypical males.
- Heterozygous females: enzyme activity overlaps with normal in up to 30–40% — enzyme assay alone CANNOT exclude Fabry in females. Genetic testing mandatory in all females with suspected Fabry.
GLA gene sequencing: Definitive diagnosis. Detects pathogenic variants; classifies as classic vs. atypical. Important for variants of uncertain significance (VUS) — functional studies or lyso-Gb3 measurement needed.
Biomarkers:
- Urine and plasma Gb3 (GL-3): Elevated in classic males. May be near-normal in atypical/late-onset and carrier females.
- Plasma lyso-Gb3: More sensitive biomarker — elevated in virtually all affected males AND in heterozygous females regardless of enzyme activity. Best biomarker for female carriers and atypical variants. Also used to monitor treatment response.
Kidney biopsy: If diagnosis uncertain or to stage nephropathy. EM shows pathognomonic zebra bodies.
Ophthalmology: Slit-lamp for cornea verticillata (confirms Gb3 storage but not specific to Fabry — also with amiodarone).
Organ-specific evaluation at diagnosis:
- eGFR, urine albumin/creatinine ratio (renal)
- ECG, echocardiogram, cardiac MRI if LVH present (cardiac)
- Brain MRI with DWI and FLAIR (cerebrovascular)
- Audiogram (hearing)
- Pain score / quality of life questionnaire
Newborn screening: Several countries now screen alpha-Gal A activity on DBS at birth (Taiwan, Italy, Austria). Cascade family screening after index case diagnosis.
9. Treatment
Enzyme Replacement Therapy (ERT): The current standard of care for Fabry patients with classic disease.
Agalsidase alfa (Replagal, Shire/Takeda): 0.2 mg/kg IV every 2 weeks. Approved in Europe, Canada, Asia. Not FDA-approved in the United States.
Agalsidase beta (Fabrazyme, Sanofi/Genzyme): 1 mg/kg IV every 2 weeks. FDA-approved and approved worldwide. Higher dose delivers more Gb3 clearance per infusion.
ERT efficacy: Reduces plasma and urine Gb3/lyso-Gb3. Stabilizes or slows renal function decline (ADVANCE trial: agalsidase beta reduced renal events 61% vs. placebo). Reduces LV mass in cardiomyopathy (with early treatment). Reduces neuropathic pain. Best outcomes with early initiation (before significant proteinuria, before eGFR <60, before significant LGE on CMR). ERT does NOT fully reverse established fibrosis/sclerosis.
Contraindications/challenges: Infusion-related reactions (IgE antibodies to agalsidase in ~50% of treated males — premedicate with antihistamine/corticosteroid; most reactions manageable). Long-term ERT antibody formation may reduce efficacy in some patients.
Migalastat (Galafold, Amicus Therapeutics): Oral pharmacological chaperone. FDA-approved 2018 for patients ≥16 years with amenable GLA variants (defined by in vitro HEK cell assay — ~35–50% of all variants are amenable). 123 mg orally every other day. Stabilizes misfolded alpha-Gal A and improves lysosomal trafficking. ATTRACT trial (Hughes et al., NEJM 2017): non-inferior to ERT for kidney function (eGFR slope); reduced LV mass; similar Gb3/lyso-Gb3 reduction. Advantages over ERT: oral, no infusion reactions, home administration. Does NOT work for null mutations (no protein to stabilize).
Symptomatic management:
- Neuropathic pain: carbamazepine (first-line for Fabry neuropathic pain — reduces frequency/severity of crises), gabapentin/pregabalin, mexiletine (severe cases); opioids avoided if possible.
- Renal protection: ACE inhibitor or ARB — reduces proteinuria and slows CKD progression (standard of care alongside ERT).
- Cardiac: antihypertensives; amiodarone (or ICD) for arrhythmia; anticoagulation for AF; heart failure management.
- Stroke prevention: antiplatelet therapy (aspirin/clopidogrel); anticoagulation for AF; blood pressure control.
- Skin: angiokeratomas — laser therapy for cosmetic improvement.
Pipeline: Venglustat (glucosylceramide synthase inhibitor, substrate reduction therapy — Phase 2/3 in Fabry); pegunigalsidase alfa (PRX-102, stabilized pegylated agalsidase beta — longer half-life, less frequent dosing, Phase 3 BALANCE trial, FDA submitted 2023); moss-derived agalsidase alfa (improved mannose-6-phosphate receptor targeting — Phase 3); gene therapy approaches (AAV-GLA) in preclinical/Phase 1.
10. Monitoring and Follow-Up
European Fabry Working Group and international guidelines recommend:
Renal: eGFR and urine albumin/creatinine ratio every 6–12 months. 24-hour urine protein if significant proteinuria. Kidney biopsy at baseline if diagnosis uncertain or to stage disease; repeat to assess ERT response debated.
Cardiac: ECG annually. Echocardiogram every 1–2 years. Cardiac MRI with gadolinium every 3–5 years (assess LGE fibrosis — strongest predictor of arrhythmia and sudden death). Holter if palpitations or syncope.
Neurological: Brain MRI at baseline and every 3–5 years (or after neurological events). Neurological exam; cognitive screening.
Pain: Validated questionnaire (Brief Pain Inventory — Fabry version). Assess pain triggers, analgesic use.
Biomarkers: Plasma lyso-Gb3 every 6–12 months (treatment response monitoring). Alpha-Gal A activity in males on ERT (to detect antibody interference).
Pregnancy: Female Fabry patients — ERT can be continued during pregnancy (risk-benefit discussion; limited data). Genetic counseling pre-conception for X-linked inheritance.
11. Research Papers (PubMed searches)
- Fabry disease alpha-galactosidase A enzyme replacement therapy
- Fabry disease migalastat ATTRACT trial GLA variants
- Fabry disease cardiomyopathy cardiac MRI late gadolinium enhancement
- Fabry disease stroke cryptogenic young adults cerebrovascular
- Fabry disease renal nephropathy proteinuria progression
- lyso-Gb3 biomarker Fabry disease females heterozygous
12. References
- Schiffmann R, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285(21):2743–2749. PMID: 11386930. https://doi.org/10.1001/jama.285.21.2743
- Banikazemi M, et al. Agalsidase-beta treatment of Fabry disease: a randomized controlled trial (ADVANCE). Ann Intern Med. 2007;146(2):77–86. PMID: 17227930. https://doi.org/10.7326/0003-4819-146-2-200701160-00148
- Hughes DA, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017;54(4):288–296. PMID: 28103158. https://doi.org/10.1136/jmedgenet-2016-104178
- Eng CM, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30(2):184–192. PMID: 17347914. https://doi.org/10.1007/s10545-006-0484-5
- Germain DP, et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016;375(6):545–555. PMID: 27509102. https://doi.org/10.1056/NEJMoa1510198
- Weidemann F, et al. Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. Circulation. 2003;108(11):1299–1301. PMID: 12939218. https://doi.org/10.1161/01.CIR.0000091253.71897.73
- Rombach SM, et al. Plasma globotriaosylsphingosine alterations in Fabry disease during enzyme replacement therapy. J Inherit Metab Dis. 2010;33(4):451–458. PMID: 20449667. https://doi.org/10.1007/s10545-010-9080-y
- Sims K, et al. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke. 2009;40(3):788–794. PMID: 19150872. https://doi.org/10.1161/STROKEAHA.108.526293
- Desnick RJ, et al. α-Galactosidase A deficiency: Fabry disease. In: Valle DL, et al., eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; 2001. Accessed at OMIM #301500.
- Nowak A, et al. Plasma lyso-Gb3: a useful biomarker for monitoring enzyme replacement therapy and evaluating the genotype-phenotype correlation in Fabry disease. Mol Genet Metab. 2017;120(1–2):57–65. PMID: 27866944. https://doi.org/10.1016/j.ymgme.2016.11.005
- Pisani A, et al. Diagnosis of Fabry disease: a clinical algorithm to identify high-risk patients. Kidney Int. 2012;82(2):174–182. PMID: 22534820. https://doi.org/10.1038/ki.2012.87
- Ortiz A, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416–427. PMID: 29530533. https://doi.org/10.1016/j.ymgme.2018.02.014
Connections
- Nephrology & Hepatology
- Chronic Kidney Disease
- IgA Nephropathy
- Alport Syndrome
- Tuberous Sclerosis Complex
- Focal Segmental Glomerulosclerosis
- Hypertrophic Cardiomyopathy
- Stroke
- Kidney Function Tests
- Urinalysis