Serine in Sphingolipid Synthesis — SPT, Myelin, and HSAN1
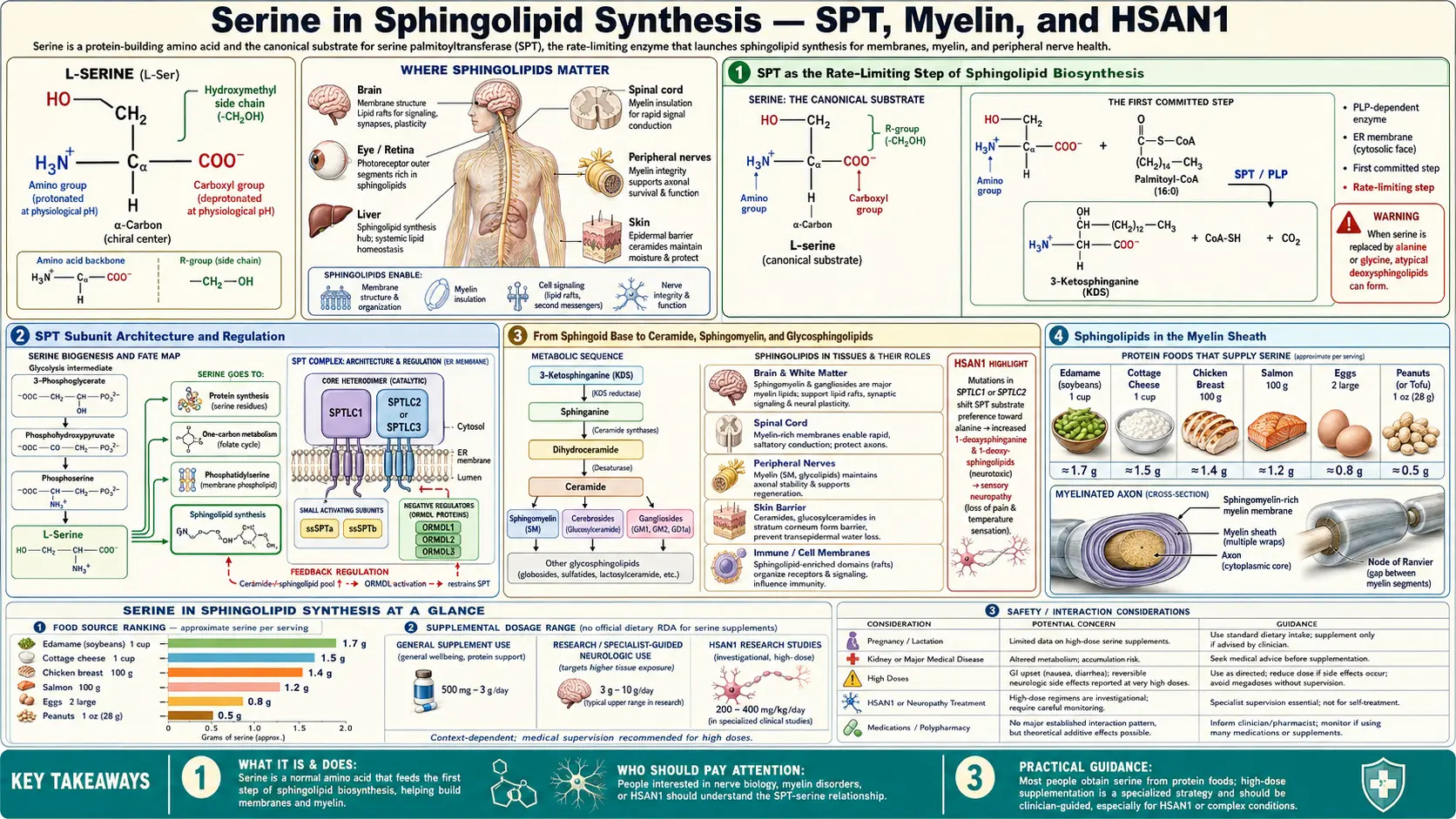
Sphingolipids are the structural backbone of the myelin sheath and a major class of membrane lipids throughout the body, accounting for roughly a third of all neural lipid mass. Their synthesis begins in a single committed reaction: serine palmitoyltransferase (SPT) condenses L-serine with palmitoyl-CoA to produce 3-ketodihydrosphingosine, the first sphingoid base. SPT is the rate-limiting step of the entire sphingolipid biosynthesis network, and serine availability directly determines its flux. Mutations in the regulatory subunit of SPT cause Hereditary Sensory and Autonomic Neuropathy type 1 (HSAN1), a progressive small-fiber sensory neuropathy in which the mutant enzyme misincorporates L-alanine in place of L-serine and produces toxic 1-deoxysphingolipids. Remarkably, oral L-serine supplementation at 200-400 mg/kg/day partially corrects the substrate-promiscuity defect, lowers plasma deoxysphingolipid levels, and slows progression of the neuropathy — one of the cleanest examples of disease-modifying amino acid therapy in modern neurology. This deep-dive walks through the SPT reaction, the sphingolipid biosynthesis network, the myelin connection, HSAN1, and the broader sphingolipid pathology that links serine metabolism to neurodegeneration, metabolic syndrome, and cancer.
Table of Contents
- SPT as the Rate-Limiting Step of Sphingolipid Biosynthesis
- SPT Subunit Architecture and Regulation
- From Sphingoid Base to Ceramide, Sphingomyelin, and Glycosphingolipids
- Sphingolipids in the Myelin Sheath
- HSAN1 — SPT Mutations and Toxic 1-Deoxysphingolipids
- L-Serine as HSAN1 Therapy — The Penno / Garofalo Trials
- Ceramides in Metabolic Syndrome and Insulin Resistance
- Lysosomal Sphingolipidoses (Gaucher, Niemann-Pick, Tay-Sachs)
- Sphingolipid Signaling in Cancer and Immunity
- L-Serine in ALS — The BMAA Hypothesis
- Key Research Papers
- Connections
- Featured Videos
SPT as the Rate-Limiting Step of Sphingolipid Biosynthesis
Serine palmitoyltransferase (SPT, EC 2.3.1.50) catalyzes the first committed reaction of sphingolipid biosynthesis:
L-serine + palmitoyl-CoA → 3-ketodihydrosphingosine + CoA + CO2
The reaction is a pyridoxal-5'-phosphate-dependent decarboxylative condensation. The PLP cofactor forms a Schiff base with the α-amino group of serine, activates the α-carbon for nucleophilic attack on the thioester carbonyl of palmitoyl-CoA, and stabilizes the carbanion intermediate that releases CO2 and produces 3-ketodihydrosphingosine. The 18-carbon product (16 from palmitoyl-CoA, 2 from serine, minus the released CO2) is the universal precursor for all sphingoid bases in mammalian cells.
SPT is the universally acknowledged rate-limiting step of de novo sphingolipid biosynthesis. Three lines of evidence support this designation:
- Enzymatic activity — SPT has the lowest specific activity (Vmax per mg protein) of any enzyme in the sphingolipid biosynthesis pathway. The downstream enzymes (3-ketosphinganine reductase, ceramide synthase, dihydroceramide desaturase) are orders of magnitude more active.
- Regulation — SPT is heavily regulated at multiple levels: transcriptional control by SREBP, post-translational regulation by the ORMDL (orosomucoid-like) proteins which inhibit SPT when sphingolipid levels are high, and direct allosteric inhibition by ceramide itself (feedback). The dense regulation pattern is characteristic of true rate-limiting enzymes.
- Pharmacological intervention — SPT inhibitors (myriocin, ISP-1, sphingofungin) potently block de novo sphingolipid synthesis without need for inhibition of any downstream enzyme.
Serine availability is one of the substrate determinants of SPT flux. In serine-replete cells, SPT operates at or near its physiological Vmax. In serine-restricted cells, SPT activity falls in proportion to serine availability, and de novo sphingolipid synthesis is reduced. The KM of SPT for serine is approximately 500 micromolar, which is in the range of typical intracellular serine concentrations, meaning that physiological serine fluctuations meaningfully shift SPT flux.
SPT Subunit Architecture and Regulation
Mammalian SPT is a heteromeric integral membrane enzyme of the endoplasmic reticulum, composed of three catalytic subunits plus regulatory partners:
- SPTLC1 (Serine Palmitoyltransferase Long Chain base subunit 1) — one of the two principal catalytic subunits. Mutations cause HSAN1 type A.
- SPTLC2 — the second principal catalytic subunit. Mutations cause HSAN1 type C.
- SPTLC3 — a tissue-restricted alternative subunit (high in placenta, kidney, intestine) that produces sphingoid bases with chain length 16 carbons rather than the canonical 18, contributing to tissue-specific sphingolipid composition.
- SPTssa, SPTssb (small subunits a and b) — required for full catalytic activity and substrate selectivity.
- ORMDL1, ORMDL2, ORMDL3 — regulatory subunits that bind SPT and inhibit its activity when cellular sphingolipid levels are sufficient. ORMDL3 is best known clinically because variants in the ORMDL3 gene are the strongest known genetic risk factor for childhood asthma, identified in GWAS studies (Moffatt 2007).
The ORMDL proteins function as the homeostatic gate of sphingolipid biosynthesis. When ceramide levels are low, ORMDLs are inhibited and SPT runs at high activity. When ceramide levels rise, ORMDLs are activated (through a complex mechanism involving direct ceramide binding), inhibit SPT, and reduce flux. This creates a tight negative-feedback loop that holds cellular sphingolipid concentrations within a narrow physiological range.
The clinical relevance of the ORMDL3 asthma association is incompletely understood but appears to involve sphingolipid signaling in airway smooth muscle and immune cells. The connection to the broader serine metabolism story is suggestive but not yet therapeutically actionable.
From Sphingoid Base to Ceramide, Sphingomyelin, and Glycosphingolipids
The 3-ketodihydrosphingosine produced by SPT is the entry point to a branching biosynthetic network that produces the entire diversity of cellular sphingolipids:
- 3-ketosphinganine reductase reduces the ketone to produce sphinganine (dihydrosphingosine)
- Ceramide synthases (CerS1-6) attach a long-chain fatty acid (usually C16 to C26) to the sphinganine amine, producing dihydroceramide. The six ceramide synthase isoforms differ in chain-length preference, producing tissue-specific ceramide species (CerS5 makes C16 ceramide; CerS1 makes C18; CerS2 makes C22-C24; CerS3 makes very long chain C26+).
- Dihydroceramide desaturase (DES1) introduces the trans-4,5 double bond to produce ceramide, the central sphingolipid intermediate.
- Ceramide is then transported to the Golgi and modified to produce:
- Sphingomyelin (SM) — by transfer of the phosphocholine head group from phosphatidylcholine, catalyzed by SM synthase. SM is the dominant sphingolipid of the plasma membrane outer leaflet and of myelin.
- Glucosylceramide and the downstream glycosphingolipid family — by sequential glycosylation. Glucosylceramide, lactosylceramide, gangliosides (GM1, GM2, GM3, GD1a, etc.), and the globoside series all derive from this branch. Gangliosides are particularly enriched in neuronal plasma membrane.
- Sphingosine-1-phosphate (S1P) — the only sphingolipid intermediate that exits the membrane and acts as a circulating signaling molecule. S1P binds five G-protein-coupled receptors (S1PR1-5) and regulates lymphocyte trafficking, vascular barrier function, and many other processes. The drug fingolimod, used in multiple sclerosis, is an S1P receptor modulator.
The total cellular sphingolipid pool is enormous — particularly in neural tissue, where sphingolipids contribute approximately 30% of total lipid mass. Each sphingolipid class has distinct functions: structural (membrane scaffold), signaling (ceramide for apoptosis, S1P for proliferation), recognition (gangliosides as cell-surface markers), and insulation (myelin sphingomyelin).
Sphingolipids in the Myelin Sheath
Myelin is the multi-lamellar membrane structure wrapped around axons by oligodendrocytes (in the central nervous system) and Schwann cells (in the peripheral nervous system). Myelin enables fast saltatory conduction of action potentials by electrically insulating the axon between nodes of Ranvier. Demyelination — loss of the myelin sheath — causes the conduction failures that underlie multiple sclerosis, Charcot-Marie-Tooth disease, and many other neurological conditions.
The lipid composition of myelin is unusual:
- 70-75% of myelin lipid is sphingolipid (compared with roughly 15-25% in most other membranes)
- Cerebrosides (galactosylceramide and its sulfated derivative sulfatide) are the most abundant single class, accounting for roughly 25% of myelin lipid mass
- Sphingomyelin contributes another 15-20%
- Cholesterol contributes about 25%
- Phospholipids (phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine) make up the rest
The high sphingolipid content gives myelin its unique mechanical and electrical properties. The trans double bond in the sphingoid base and the saturated long-chain fatty acid in the ceramide create tightly packed bilayers with high transition temperatures and low water content — ideal for electrical insulation. The galactosylceramide and sulfatide head groups participate in trans-cellular interactions that hold adjacent myelin wraps together (the major dense line of compact myelin).
The synthesis of all this sphingolipid requires substantial serine flux. Each ceramide molecule requires one serine via SPT. A typical adult brain contains on the order of 30 to 40 grams of sphingolipid, and the steady-state turnover of myelin sphingolipid consumes serine continuously. During the developmental period of active myelination (peak in the first 2 years of life, continuing through young adulthood), serine demand for myelin synthesis is particularly high. Inborn errors of serine biosynthesis (PHGDH, PSAT1, or PSPH deficiency) present in infancy with severe microcephaly and white matter abnormalities, reflecting the dependence of myelination on serine supply.
HSAN1 — SPT Mutations and Toxic 1-Deoxysphingolipids
Hereditary Sensory and Autonomic Neuropathy type 1 (HSAN1, OMIM 162400) is a dominantly inherited progressive sensory neuropathy that begins in the second to fourth decade of life with loss of pain and temperature sensation in the feet, followed by ulceration, Charcot joints, and amputation. Motor function is relatively preserved early, but progressive weakness develops in advanced disease. The condition was characterized clinically in the 1970s and the genetic basis identified in 2001 by Dawkins et al., who reported missense mutations in SPTLC1 (the principal catalytic subunit of SPT) as the cause.
The mutations are concentrated at specific residues (most commonly Cys133Trp, Cys133Tyr, Val144Asp) that lie in or near the active site. The genetics was initially confusing because the mutations are autosomal dominant rather than recessive — expected if the mutations were simple loss-of-function. Loss of one copy of SPTLC1 would be expected to be tolerated (half-normal enzyme activity is sufficient for sphingolipid synthesis), but the dominant inheritance suggested a gain-of-function mechanism.
The mechanism was clarified in a series of papers by Hornemann, Penno, and Eckardt between 2009 and 2012. The HSAN1 mutations change the substrate specificity of SPT — the mutant enzyme can use L-alanine in place of L-serine as the α-amino acid substrate. The alanine-substituted product is 1-deoxysphinganine, which lacks the hydroxyl group of normal sphinganine. 1-deoxysphinganine can be elongated and acylated by downstream enzymes to produce 1-deoxyceramides and 1-deoxysphingosines, an entire family of "deoxy" sphingolipids that cannot be processed by the normal sphingolipid degradation enzymes (sphingosine kinase, ceramidase) because the missing hydroxyl is the substrate handle for those enzymes.
The deoxysphingolipids accumulate in plasma and in tissues, particularly in sensory neurons of the dorsal root ganglia. They are directly neurotoxic — cell culture studies show that exposure to 1-deoxysphinganine at the concentrations measured in HSAN1 patient plasma kills sensory neurons within days. The toxicity drives the progressive sensory neuropathy that is the clinical hallmark of HSAN1.
HSAN1 is now recognized as a paradigmatic gain-of-function disease where the mutant enzyme produces a toxic metabolite that the normal enzyme does not. Similar gain-of-function neuropathy mechanisms have been recognized in other disorders (e.g., GM1 gangliosidosis sialic acid accumulation, deoxyguanosine kinase deficiency, several mitochondrial DNA depletion syndromes), and HSAN1 is the classic example of the principle.
L-Serine as HSAN1 Therapy — The Penno / Garofalo Trials
The mechanism of HSAN1 — mutant SPT using alanine when it cannot find enough serine — immediately suggests a therapeutic strategy: if extracellular serine concentrations are raised high enough, the mutant enzyme's substrate competition will favor serine and the production of toxic 1-deoxysphingolipids will fall.
This was tested first in the SPTLC1-C133W mouse model (Eichler 2009) and then in HSAN1 patients. Garofalo and Penno reported in 2011 (PNAS, JCI) that oral L-serine supplementation at approximately 400 mg/kg/day — roughly 30 grams/day for an adult — reduced plasma 1-deoxysphingoid base levels by 50-70% in HSAN1 patients within weeks of starting supplementation. A subsequent 2019 randomized controlled trial (Fridman et al., Annals of Neurology) enrolled 18 HSAN1 patients in a placebo-controlled crossover design with 6 months on each arm. The L-serine arm showed:
- Significant reduction in plasma 1-deoxysphinganine and 1-deoxysphingosine
- Improvement on the Charcot-Marie-Tooth Neuropathy Score (a validated clinical neuropathy outcome measure)
- Excellent safety profile — the very large doses (30 g/day) were well tolerated without significant adverse effects
L-serine supplementation is now standard of care for HSAN1, recommended by international neurology consensus guidelines. The supplementation does not reverse damage that has already occurred but slows or arrests further progression. It is one of the clearest examples in modern neurology of a disease-modifying amino acid therapy — the mechanistic reasoning is precise, the biochemical effect is measurable in plasma, and the clinical benefit is real.
The HSAN1 story is also a beautiful illustration of how understanding a single enzyme's substrate specificity can lead directly to a rational therapy. The reasoning from "mutation alters substrate selectivity" to "compete with toxic alternate substrate by flooding with correct substrate" is the kind of clean mechanistic logic that more often appears in textbooks than in clinical practice.
Ceramides in Metabolic Syndrome and Insulin Resistance
Beyond myelin and HSAN1, sphingolipids have emerged in the 2010s as central players in metabolic disease. Ceramides — the central intermediate in sphingolipid metabolism — act as bioactive signaling molecules that directly impair insulin sensitivity in muscle, liver, and adipose tissue.
Multiple mechanisms link ceramide to insulin resistance:
- Akt inhibition — ceramide activates protein phosphatase 2A (PP2A), which dephosphorylates and inactivates Akt, the central kinase of insulin signaling. The result is reduced insulin-stimulated glucose uptake in muscle and reduced insulin-mediated suppression of hepatic glucose output.
- PKCζ activation — ceramide also activates atypical protein kinase C zeta, which phosphorylates Akt at an inhibitory site, further suppressing insulin signaling.
- Mitochondrial dysfunction — ceramide accumulation in mitochondria impairs electron transport chain function and reduces fatty acid oxidation, contributing to lipid accumulation that drives further ceramide synthesis (a vicious cycle).
- Inflammation — specific ceramide species (particularly C16 and C18 ceramides) drive TLR4 signaling and inflammatory cytokine production in adipose tissue macrophages.
Plasma ceramide concentrations are strongly associated with insulin resistance, type 2 diabetes risk, cardiovascular events, and all-cause mortality in human cohort studies. The Mayo Clinic Ceramide Score (Cer16, Cer18, Cer24, Cer24:1) is a validated clinical lab test for cardiovascular risk stratification that outperforms LDL cholesterol in some populations.
The therapeutic implications are still being worked out. Reducing ceramide accumulation through SPT inhibition (myriocin), ceramide synthase inhibition, or direct lipid metabolism modulation has shown preclinical promise in metabolic disease models but has not yet produced approved drugs. Dietary interventions (Mediterranean diet, omega-3 fatty acids, weight loss) reduce plasma ceramide concentrations, which is part of the metabolic benefit of these approaches.
For more on metabolic syndrome and insulin resistance, see our Metabolic Syndrome page and Insulin Resistance page.
Lysosomal Sphingolipidoses (Gaucher, Niemann-Pick, Tay-Sachs)
Loss-of-function mutations in the lysosomal sphingolipid catabolic enzymes cause the lysosomal storage disorders collectively called sphingolipidoses. These are rare but well-characterized inherited disorders that have shaped the understanding of sphingolipid biology:
- Gaucher disease (glucocerebrosidase deficiency) — glucocerebroside accumulation in liver, spleen, bone marrow. The most common sphingolipidosis, with effective enzyme replacement therapy (imiglucerase, taliglucerase) and substrate reduction therapy (miglustat, eliglustat).
- Niemann-Pick disease types A and B (acid sphingomyelinase deficiency) — sphingomyelin accumulation. Type A causes fatal infantile neurodegeneration; type B is a milder visceral form.
- Tay-Sachs disease and Sandhoff disease (hexosaminidase deficiency) — GM2 ganglioside accumulation in neurons. Tay-Sachs causes fatal infantile neurodegeneration.
- Fabry disease (alpha-galactosidase deficiency) — globotriaosylceramide accumulation in vascular endothelium, kidney, heart, peripheral nerves.
- Krabbe disease (galactocerebrosidase deficiency) — galactosylceramide accumulation; fatal infantile neurodegeneration.
- Metachromatic leukodystrophy (arylsulfatase A deficiency) — sulfatide accumulation; progressive demyelination.
These disorders are not caused by serine metabolism abnormalities — they are caused by failure to break down the sphingolipid products that serine helped build. But they are part of the broader sphingolipid biology framework, and understanding them illuminates the normal homeostasis of sphingolipid turnover. Each is now an established target of enzyme replacement therapy, substrate reduction therapy, or chaperone therapy, and several have benefited from gene therapy approaches in recent years.
Sphingolipid Signaling in Cancer and Immunity
Beyond their structural roles, ceramide and sphingosine-1-phosphate (S1P) are bioactive signaling lipids with opposing effects on cell fate:
- Ceramide promotes apoptosis (programmed cell death), cell-cycle arrest, and senescence. Many chemotherapy drugs (doxorubicin, etoposide, radiation, Fas ligand) work in part by triggering ceramide accumulation in tumor cells. Inducing ceramide accumulation is therefore one of the strategies in cancer therapy.
- S1P promotes proliferation, survival, angiogenesis, and immune cell trafficking. S1P signaling supports tumor growth and metastasis, and inhibitors of S1P production (sphingosine kinase inhibitors) or S1P receptors (fingolimod, ozanimod, ponesimod) are explored as cancer adjuvants.
The ratio of ceramide to S1P (the "sphingolipid rheostat") regulates the balance between cell death and survival, and shifting this rheostat is an active area of cancer drug development. The rheostat is also relevant in immunology — fingolimod, the first S1P-receptor modulator approved for multiple sclerosis, works by trapping lymphocytes in lymph nodes (preventing their migration to the CNS) through internalization of the S1P1 receptor.
Serine supply influences the entire sphingolipid network through SPT, and therefore indirectly modulates the ceramide-S1P balance. The relevance for cancer biology overlaps with the serine-and-glycine restriction strategy discussed in our Glycine Source deep-dive — serine restriction reduces both one-carbon metabolism and sphingolipid biosynthesis, both of which support tumor growth.
L-Serine in ALS — The BMAA Hypothesis
L-serine has been promoted in alternative medicine as a treatment for amyotrophic lateral sclerosis (ALS) based on a hypothesis advanced by Paul Cox and colleagues at the Institute for Ethnomedicine. The proposed mechanism: the cyanobacterial non-protein amino acid β-N-methylamino-L-alanine (BMAA) can be misincorporated into proteins in place of L-serine, producing misfolded proteins that drive neurodegeneration. Oral L-serine supplementation, in this hypothesis, competes with BMAA for protein incorporation and reduces the misfolded protein burden.
The BMAA-ALS hypothesis remains controversial. A small Phase IIa trial of L-serine at 15 g/day in ALS reported safety and a small effect on disease progression, but the trial was not powered for efficacy and the result has not been replicated in a larger randomized trial. Mechanistically, the BMAA-misincorporation hypothesis is debated — whether BMAA is actually misincorporated into proteins at biologically relevant rates remains contested, and the population epidemiology linking BMAA exposure to ALS clusters is mixed.
Importantly, the D-serine literature (see our D-Serine and NMDA deep-dive) shows that D-serine accumulates pathologically in ALS spinal cord and drives motor neuron excitotoxicity (Sasabe 2007). The relationship between L-serine supplementation and the spinal-cord D-serine accumulation is not fully worked out — in theory, providing more L-serine substrate could either help (if serine racemase is downregulated and the issue is reduced D-serine clearance) or hurt (if it provides more substrate for the elevated serine racemase activity in ALS-affected tissue).
The practical conclusion: high-dose L-serine for ALS should be undertaken only within formal clinical trials or with neurology supervision, and the mechanism is more complex than the BMAA hypothesis suggests. The HSAN1 indication for L-serine is, by contrast, mechanistically rigorous and clinically established. For broader ALS information, see our ALS page.
Key Research Papers
- Dawkins JL et al. (2001). Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nature Genetics. — PubMed
- Penno A et al. (2010). Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. Journal of Biological Chemistry. — PubMed
- Garofalo K et al. (2011). Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. Journal of Clinical Investigation. — PubMed
- Fridman V et al. (2019). Randomized trial of l-serine in patients with hereditary sensory and autonomic neuropathy type 1. Annals of Neurology. — PubMed
- Hannun YA, Obeid LM (2018). Sphingolipids and their metabolism in physiology and disease. Nature Reviews Molecular Cell Biology. — PubMed
- Breslow DK, Weissman JS (2010). Membranes in balance: mechanisms of sphingolipid homeostasis. Molecular Cell. — PubMed
- Summers SA, Chaurasia B, Holland WL (2019). Metabolic messengers: ceramides. Nature Metabolism. — PubMed
- Hornemann T et al. (2009). The SPTLC3 subunit of serine palmitoyltransferase generates short chain sphingoid bases. Journal of Biological Chemistry. — PubMed
- Cox PA et al. (2016). Dietary exposure to an environmental toxin triggers neurofibrillary tangles and amyloid deposits in the brain. Proceedings of the Royal Society B. — PubMed
- Eichler FS et al. (2009). Overexpression of the wild-type SPT1 subunit lowers desoxysphingolipid levels and rescues the phenotype of HSAN1. Journal of Neuroscience. — PubMed
- Moffatt MF et al. (2007). Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. — PubMed
- Chaurasia B, Summers SA (2015). Ceramides — lipotoxic inducers of metabolic disorders. Trends in Endocrinology and Metabolism. — PubMed
PubMed Topic Searches
- PubMed: SPT and sphingolipid synthesis
- PubMed: HSAN1 / SPTLC mutations
- PubMed: L-serine for HSAN1
- PubMed: Myelin sphingolipids
- PubMed: Ceramides and metabolic disease
Connections
- Serine Overview
- Serine Benefits Hub
- Phosphatidylserine and Brain
- D-Serine and NMDA
- Glycine Source (SHMT)
- Alanine (HSAN1 Competitor)
- Vitamin B6 (SPT Cofactor)
- ALS
- Multiple Sclerosis
- Metabolic Syndrome
- Insulin Resistance
- Asthma (ORMDL3 Link)
- Type 2 Diabetes
- Omega-3 Fatty Acids
- All Amino Acids