Vitamin B4 (Adenine) and Purine Metabolism
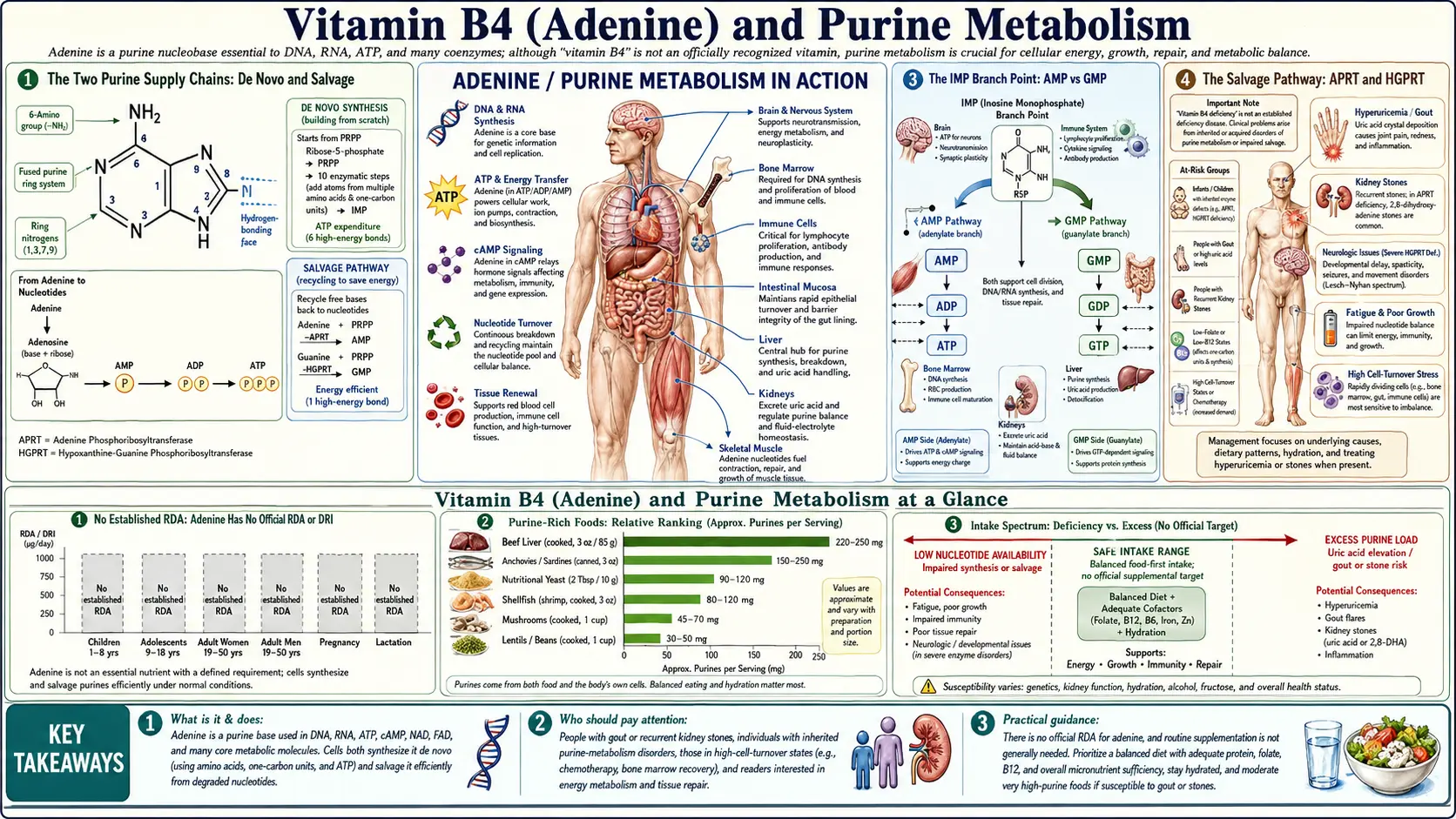
Purine metabolism is the biochemical machinery that builds, recycles, and ultimately disposes of adenine and guanine — the two purine bases of DNA, RNA, ATP, GTP, NAD, and dozens of cofactors. The human body operates two complementary supply chains: a costly de novo pathway that builds the purine ring from scratch over ten enzymatic steps (consuming six ATP for every IMP produced), and a thrifty salvage pathway that recycles purine bases liberated by nucleic acid turnover. In healthy adults, salvage accounts for roughly 90% of daily purine supply; de novo synthesis fills the remaining 10% and ramps up in rapidly proliferating tissues such as bone marrow, gut crypts, and embryonic tissue. When salvage breaks (as in Lesch-Nyhan syndrome) or when overproduction occurs (as in tumor lysis syndrome and certain forms of gout), the clinical consequences are dramatic, irreversible, and instructive.
Table of Contents
- The Two Purine Supply Chains: De Novo and Salvage
- The De Novo Pathway: Ten Steps from PRPP to IMP
- The IMP Branch Point: AMP vs GMP
- The Salvage Pathway: APRT and HGPRT
- Pathway Regulation and Feedback Inhibition
- Catabolism: From Adenine to Uric Acid
- Clinical Disorders of Purine Metabolism
- Drug Targets in Purine Metabolism
- Dietary Purines and Endogenous Synthesis
- Key Research Papers
- Connections
- Featured Videos
The Two Purine Supply Chains: De Novo and Salvage
Every cell faces a continuous demand for purine nucleotides. A typical proliferating human cell requires on the order of 1010 purine residues per cell division to populate new DNA, RNA, ATP pools, and cofactors. Resting cells have a more modest but still substantial baseline turnover — RNA is constantly transcribed and degraded, ATP is constantly hydrolyzed and resynthesized, and DNA repair operates around the clock.
Two metabolic pathways meet that demand. The de novo synthesis pathway builds the purine ring atom by atom from small precursors: glycine donates two carbons and one nitrogen; aspartate donates one nitrogen; glutamine donates two nitrogens; and one-carbon units carried by tetrahydrofolate (THF) donate two ring carbons. The pathway costs six ATP equivalents per inosine monophosphate (IMP) produced, plus the glycine, glutamine, and aspartate consumed. This is not a cheap operation.
The salvage pathway reuses free purine bases (adenine, hypoxanthine, guanine) that have been liberated by nucleotide breakdown. A single enzymatic step combines the free base with PRPP (5-phosphoribosyl-1-pyrophosphate) to regenerate the nucleotide. Salvage costs roughly one ATP per nucleotide reformed — six times cheaper than de novo synthesis. The trade-off is that salvage only works if the cell has access to free purine bases either from its own turnover or from dietary/blood sources.
In practice, the two pathways are reciprocally regulated and operate together. Tissue type sets the balance. Hepatocytes and rapidly proliferating cells use mostly de novo. Mature lymphocytes, neurons, and erythrocytes rely heavily on salvage because their de novo capacity is limited. This compartmentalization is why some purine disorders (Lesch-Nyhan, ADA-SCID) preferentially damage specific cell populations rather than the whole organism.
The De Novo Pathway: Ten Steps from PRPP to IMP
The de novo pathway begins with 5-phosphoribosyl-1-pyrophosphate (PRPP), itself synthesized by PRPP synthetase from ribose-5-phosphate and ATP. PRPP is the activated sugar-phosphate scaffold on which the purine ring will be assembled, atom by atom, in a metabolic synthesis that resembles building a house on a foundation pad. The ten enzymatic steps from PRPP to IMP are catalyzed by enzymes condensed (in vertebrates) onto only a handful of large multi-functional polypeptides — TRIFGART, PAICS, ATIC, GART, and others — that channel intermediates without releasing them to bulk solvent.
The committed step is the second reaction: glutamine PRPP amidotransferase (GPAT, also known as PPAT) transfers the amide nitrogen of glutamine to PRPP, displacing pyrophosphate and creating 5-phosphoribosylamine. This is the rate-limiting and feedback-controlled step. AMP, ADP, GMP, and GDP all inhibit GPAT allosterically, so high purine nucleotide concentrations slow further synthesis. The committed step also depletes intracellular glutamine, which is why purine synthesis is highly responsive to glutamine availability and why rapidly proliferating tumors often display "glutamine addiction."
The subsequent eight steps add the remaining ring atoms in sequence. Glycine contributes a full two-carbon, one-nitrogen unit. The N5,N10-methenyl- and N10-formyl-THF derivatives of folate contribute two ring carbons through formyltransferase reactions — the explanation for why antifolate drugs (methotrexate, pemetrexed) cripple purine synthesis. Aspartate contributes one nitrogen through an unusual two-step condensation-elimination via adenylosuccinate intermediates. The final product, inosine monophosphate (IMP), contains hypoxanthine as the nitrogenous base.
IMP is the universal purine precursor. It is then channeled into either AMP synthesis (via adenylosuccinate synthetase, with GTP as energy source) or GMP synthesis (via IMP dehydrogenase + GMP synthetase, with NAD+ and ATP). The crossover energetics are deliberate: AMP synthesis costs GTP, and GMP synthesis costs ATP — a reciprocal-cost design that maintains the AMP/GMP ratio when both purines are needed in similar quantities.
The IMP Branch Point: AMP vs GMP
The IMP-to-AMP branch is catalyzed in two steps: adenylosuccinate synthetase adds aspartate's amino group across the 6-position (consuming GTP), and adenylosuccinate lyase eliminates fumarate to yield AMP. Fumarate enters the citric acid cycle, recycling carbon back into central metabolism.
The IMP-to-GMP branch is also two steps: IMP dehydrogenase (IMPDH) oxidizes IMP at the 2-position to xanthosine monophosphate (XMP), consuming NAD+; GMP synthetase then aminates XMP at the 2-position using glutamine, consuming ATP. IMPDH is the rate-limiting step for GMP synthesis and is the molecular target of mycophenolate mofetil (CellCept), a widely used immunosuppressant in transplantation. Lymphocytes are exquisitely sensitive to IMPDH inhibition because they rely heavily on de novo GMP synthesis — their salvage capacity for guanine is limited.
Once AMP and GMP are made, they are sequentially phosphorylated by adenylate kinase and nucleoside-diphosphate kinase to produce ADP and ATP, GDP and GTP. The triphosphate forms are the active species used in nucleic acid synthesis (dATP and dGTP, after ribonucleotide reductase action), energy currency (ATP), and signaling (GTP for G-proteins, cAMP and cGMP as second messengers).
The Salvage Pathway: APRT and HGPRT
The salvage pathway is conceptually simpler than de novo synthesis: a single enzyme attaches a pre-formed free base (adenine, hypoxanthine, or guanine) to PRPP, regenerating the corresponding nucleotide. Two enzymes handle the three substrates:
- APRT (adenine phosphoribosyltransferase) salvages adenine to AMP. Adenine + PRPP → AMP + PPi.
- HGPRT (hypoxanthine-guanine phosphoribosyltransferase, also written HPRT) salvages both hypoxanthine to IMP and guanine to GMP. The single enzyme handles both purine bases.
The substrates — adenine, hypoxanthine, guanine — arise continuously from RNA and DNA turnover, from purine nucleotide catabolism, and (in small quantities) from dietary nucleic acid breakdown in the gut. Most cells do not absorb intact dietary nucleotides; the gut lumen and enterocyte break ingested nucleic acids down to the free base, which then enters the bloodstream and is salvaged by tissues with high HGPRT or APRT activity.
The energetic logic is striking. De novo IMP synthesis from PRPP costs roughly six ATP, plus the consumption of one glutamine (with its own metabolic cost) and one glycine. Salvage of hypoxanthine to IMP costs one ATP (used by PRPP synthetase to make PRPP). The roughly six-fold cost advantage explains why selection has retained salvage enzymes even in the face of de novo capability — salvage frees up energy and amino-acid carbon for other purposes.
Pathway Regulation and Feedback Inhibition
Purine biosynthesis is tightly regulated at multiple control points to match supply with demand and to avoid wasteful overproduction.
PRPP synthetase is regulated by ADP and GDP allosterically. When the cell's nucleotide pools are full, PRPP synthesis slows, throttling both de novo and salvage pathway capacity.
Glutamine PRPP amidotransferase (GPAT), the committed step of de novo synthesis, is allosterically inhibited by AMP, ADP, GMP, and GDP. This is the classic end-product feedback loop. GPAT also has cooperative substrate binding for PRPP, so it switches on sharply when PRPP rises above threshold and switches off sharply when it falls below.
Adenylosuccinate synthetase (IMP → AMP) is inhibited by AMP, providing branch-specific feedback control.
IMP dehydrogenase (IMP → XMP) is inhibited by GMP, providing reciprocal branch-specific control.
The reciprocal regulation ensures that overproduction of either AMP or GMP slows further synthesis at the relevant branch while leaving the other branch intact. This homeostatic design maintains the AMP/GMP ratio at roughly 4:1 across most cell types, with deviations primarily reflecting tissue-specific demand (myocardium needs more ATP; neurons need more GTP for vesicle fusion).
Catabolism: From Adenine to Uric Acid
What goes up must come down. Purine nucleotides do not last forever — mRNA is degraded after translation, ATP is hydrolyzed in muscle contraction, DNA undergoes constant repair-and-replace cycles, and cells die and are recycled. The breakdown pathway is shared for both adenine-containing and guanine-containing nucleotides and converges on a single end product: uric acid.
Adenine-containing nucleotides (AMP, ADP, ATP) are deaminated by adenosine deaminase (ADA) or by AMP deaminase to give inosine and IMP, respectively. IMP/inosine are then cleaved by purine nucleoside phosphorylase (PNP) to free hypoxanthine. Guanine-containing nucleotides (GMP, GDP, GTP) are cleaved by PNP to free guanine, which is then deaminated by guanase to xanthine.
The final two oxidative steps are catalyzed by xanthine oxidase (XO; also called xanthine dehydrogenase in its reduced form): hypoxanthine is oxidized to xanthine, and xanthine is further oxidized to uric acid. Uric acid in humans is the end product because we lack functional uricase (the enzyme that, in most other mammals, would further degrade uric acid to the much more soluble allantoin). The uricase gene became a non-functional pseudogene in the hominoid lineage approximately 15 million years ago, leaving humans with relatively high blood uric acid concentrations — the price we pay for whatever evolutionary advantage the uricase loss provided.
Uric acid is poorly water-soluble at physiologic pH and tends to crystallize as monosodium urate when concentrations exceed roughly 6.8 mg/dL. Crystallization in synovial fluid produces gouty arthritis; crystallization in the kidney produces uric acid stones and uric acid nephropathy. The therapeutic target xanthine oxidase is inhibited by allopurinol and febuxostat, both used to manage chronic hyperuricemia and gout.
Clinical Disorders of Purine Metabolism
Several rare inherited disorders illuminate what happens when individual enzymes of purine metabolism break. Each provides a natural experiment that has informed our understanding of pathway logic.
Lesch-Nyhan syndrome is X-linked, caused by mutations in HGPRT. Affected boys cannot salvage hypoxanthine or guanine, so they overproduce uric acid (via the upstream pathway pushing more substrate through xanthine oxidase) and develop severe early-onset gout, urate nephropathy, dystonia, intellectual disability, and the pathognomonic self-injurious behavior (lip biting, finger biting). The neurological phenotype is not explained by uric acid — the brain is largely uric-acid-blind — but by failure of salvage in basal-ganglia neurons, which apparently depend heavily on HGPRT-mediated guanine salvage for normal dopaminergic function.
Adenine phosphoribosyltransferase (APRT) deficiency is autosomal recessive and causes 2,8-dihydroxyadenine urolithiasis. Without APRT, free adenine accumulates and is oxidized by xanthine oxidase to the very poorly soluble 2,8-dihydroxyadenine, which precipitates in the kidney as crystals, sand, and stones. The crystals are radiolucent and can be mistaken for uric acid stones — the distinction matters because allopurinol prevents the dihydroxyadenine formation and is curative, while a low-purine diet alone is not.
Adenosine deaminase (ADA) deficiency produces severe combined immunodeficiency (ADA-SCID). The accumulated deoxyadenosine is toxic to developing T and B lymphocytes through depletion of S-adenosylhomocysteine hydrolase and elevation of dATP, which dysregulates ribonucleotide reductase. Without enzyme replacement therapy (PEG-ADA), bone-marrow transplant, or gene therapy, affected infants die of infection in the first year of life. ADA-SCID was historically the first human disease successfully treated by gene therapy (Cavazzana-Calvo and colleagues, 1990s).
Purine nucleoside phosphorylase (PNP) deficiency produces a milder T-cell selective immunodeficiency through accumulation of deoxyguanosine and its conversion to dGTP, which similarly poisons proliferating T lymphocytes via ribonucleotide reductase inhibition.
Common gout is not a single-enzyme disorder. Most primary gout reflects renal underexcretion of uric acid, with diet, alcohol, fructose intake, and genetic polymorphisms in urate transporters (URAT1, GLUT9, ABCG2) as risk modifiers. A minority of gout patients overproduce uric acid through partial HGPRT deficiency or PRPP synthetase overactivity.
Drug Targets in Purine Metabolism
The purine pathway is one of the most heavily drugged metabolic networks in medicine, with applications spanning cancer, transplantation, autoimmunity, and antiviral therapy.
Methotrexate inhibits dihydrofolate reductase, depleting the reduced-folate cofactors required for one-carbon transfer steps in de novo purine synthesis. Used in acute lymphoblastic leukemia, rheumatoid arthritis, psoriasis, and ectopic pregnancy management.
6-mercaptopurine and 6-thioguanine are purine analogs that mimic hypoxanthine and guanine. After salvage by HGPRT, the resulting thio-IMP and thio-GMP feedback-inhibit GPAT and incorporate into DNA, killing proliferating cells. Used in acute lymphoblastic leukemia.
Azathioprine is a 6-mercaptopurine prodrug used as an immunosuppressant in IBD, lupus, and transplantation. Patients with thiopurine methyltransferase (TPMT) deficiency cannot detoxify the active metabolites and develop severe myelosuppression at standard doses — TPMT genotyping before initiation is now standard of care.
Mycophenolate mofetil inhibits IMP dehydrogenase, selectively depleting GMP in lymphocytes that lack robust salvage capacity. Workhorse immunosuppressant in solid-organ transplantation.
Allopurinol and febuxostat inhibit xanthine oxidase, lowering uric acid production and treating chronic gout. Allopurinol also potentiates 6-mercaptopurine and azathioprine (xanthine oxidase normally degrades them), requiring dose reduction.
Acyclovir, ganciclovir, tenofovir, and other antiviral nucleoside analogs are purine mimics that, once phosphorylated by viral enzymes, are incorporated into viral DNA and terminate elongation. The selectivity comes from the preferential phosphorylation by viral thymidine kinase (acyclovir) or selective uptake into infected cells.
Dietary Purines and Endogenous Synthesis
Healthy adults synthesize and salvage all the purines they need without depending on diet. A typical Western diet contributes 100–300 mg of preformed purines per day, primarily from meat and fish; vegetarian diets contribute less but include yeast extract, legumes, and some vegetables (asparagus, spinach, mushrooms) with moderate purine content. Dietary purines are mostly broken down in the gut to free bases, absorbed across the intestinal epithelium, and salvaged or excreted as uric acid.
For most people, dietary purine intake has only a modest effect on serum uric acid — perhaps 1–2 mg/dL difference between a high-purine and low-purine diet. The reason is that endogenous synthesis dwarfs dietary input in absolute terms, and the body upregulates de novo synthesis when dietary salvage substrate is low.
In gout patients, the marginal effect of diet still matters because uric acid solubility has a hard threshold around 6.8 mg/dL. Pushing a patient from 7.5 mg/dL down to 6.5 mg/dL by avoiding high-purine foods, alcohol (particularly beer, which contains both purines and ethanol that competes with uric acid for renal excretion), and high-fructose corn syrup can substantially reduce flare frequency. The 2020 ACR Gout Guidelines emphasize that diet is adjunctive to urate-lowering pharmacotherapy, not a substitute.
In tumor lysis syndrome, the explosive release of intracellular nucleic acids during chemotherapy-induced cell death can overwhelm the body's capacity to excrete uric acid, causing acute uric acid nephropathy and renal failure. Allopurinol prophylaxis or, for high-risk patients, recombinant uricase (rasburicase) converts uric acid to soluble allantoin, restoring renal handling and preventing renal failure.
In total parenteral nutrition (TPN) and infant formula, nucleotide supplementation has been studied as a conditionally essential nutrient. Premature infants, infants with short bowel syndrome, and critically ill patients with high cell-turnover demand may benefit from exogenous nucleotides when endogenous synthesis cannot keep up. Standard infant formulas have included nucleotide supplementation since the 1990s on this rationale.
Key Research Papers
- Buchanan JM, de novo purine biosynthesis from glycine, formate, and aspartate — PubMed: Buchanan de novo pathway
- Seegmiller JE and Rosenbloom FM, HGPRT enzyme deficiency in Lesch-Nyhan syndrome (1967) — PubMed: Seegmiller HGPRT 1967
- Wyngaarden JB and Kelley WN, classic purine metabolism reviews — PubMed: Wyngaarden Kelley
- Cavazzana-Calvo M et al., gene therapy for ADA-SCID — PubMed: ADA-SCID gene therapy
- Allison AC, mycophenolate mofetil mechanism via IMP dehydrogenase inhibition — PubMed: Allison mycophenolate
- Elion GB, design of allopurinol and 6-mercaptopurine (Nobel work) — PubMed: Elion drug design
- Becker MA, gout pathophysiology and uric acid homeostasis — PubMed: Becker gout review
- Lesch M, Nyhan WL, original 1964 description of the syndrome — PubMed: Lesch-Nyhan original 1964
- Bridges AJ, allopurinol-induced hypersensitivity syndrome (HLA-B*58:01 association) — PubMed: Allopurinol HLA-B*58:01
- Lipkowitz MS, urate transporters URAT1, GLUT9, ABCG2 in renal handling of uric acid — PubMed: Urate transporters
- FitzGerald JD, 2020 American College of Rheumatology guidelines for management of gout — PubMed: ACR gout guidelines 2020
- Coiffier B, tumor lysis syndrome and rasburicase clinical efficacy — PubMed: Coiffier TLS
Connections
- Vitamin B4 Benefits (Hub)
- Vitamin B4 (Main Page)
- Cellular Energy and ATP
- DNA and RNA Synthesis
- Modern Status and Food Sources
- Vitamin B9 (Folate — one-carbon donor)
- Vitamin B3 (NAD — oxidant in IMPDH)
- Gout
- Kidney Stones
- Uric Acid Test
- Glutamine (purine ring nitrogen donor)
- Glycine (purine ring carbon donor)
- Aspartate (purine ring nitrogen donor)
- All Vitamins