Mitochondrial Health and Oxidative Stress
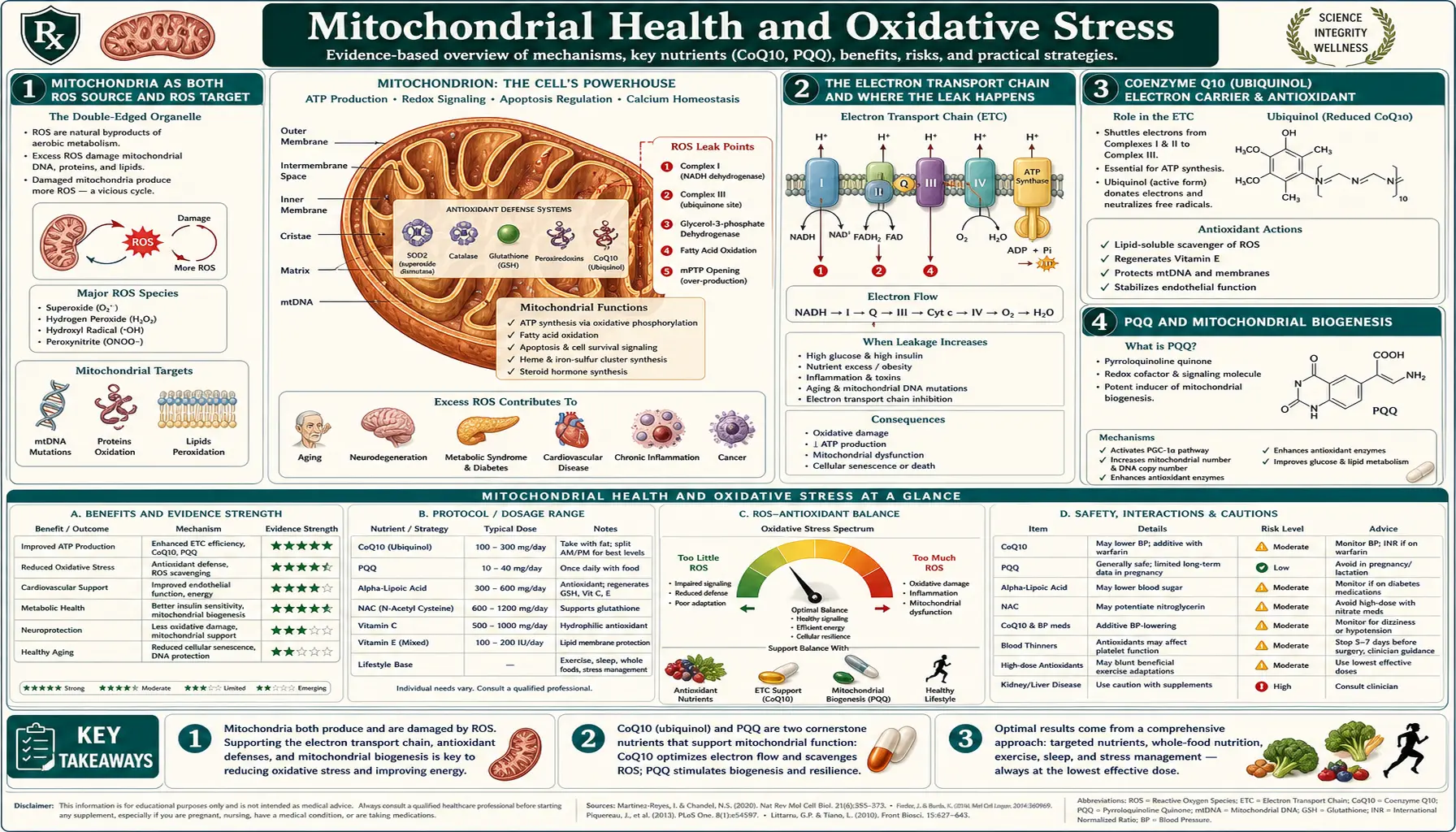
Mitochondria are the dominant source of reactive oxygen species in human cells — approximately 0.2 to 2 percent of the oxygen they consume escapes the electron transport chain as superoxide. They are also the dominant target of oxidative damage, because mitochondrial DNA sits unprotected by histones, just nanometers from the leaky electron transport chain. The result is a feedback loop where mitochondrial dysfunction generates more ROS, which damages more mitochondria. This loop is now recognized as a central feature of nearly every chronic disease and the aging process itself — the Harman free-radical theory of aging, while incomplete, captures something real about mitochondrial decline. Supporting mitochondrial health is therefore not just one antioxidant strategy among many; it is the strategy that targets ROS production at its source rather than mopping up afterward. This deep-dive walks through the electron transport chain's role as both ROS producer and target, the supplements with the best evidence (CoQ10, PQQ, NAD+ precursors, alpha-lipoic acid), the indispensable role of exercise as the most potent mitochondrial biogenesis stimulus, and how mitochondrial dysfunction unifies the metabolic-neurodegenerative-aging disease cluster.
Table of Contents
- Mitochondria as Both ROS Source and ROS Target
- The Electron Transport Chain and Where the Leak Happens
- Coenzyme Q10 (Ubiquinol) — Electron Carrier and Antioxidant
- PQQ and Mitochondrial Biogenesis
- NAD+ Decline with Aging and NMN/NR Supplementation
- Alpha-Lipoic Acid as the Mitochondrial Cofactor
- Exercise — The Most Potent Mitochondrial Biogenesis Stimulus
- Mitophagy: Removing Damaged Mitochondria
- Mitochondrial Dysfunction as Unifying Disease Mechanism
- Cautions and Practical Stack
- Key Research Papers
- Connections
- Featured Videos
Mitochondria as Both ROS Source and ROS Target
Every cell in the body except mature red blood cells contains mitochondria — from a handful in lymphocytes to thousands in cardiomyocytes and hepatocytes. They produce the ATP that powers virtually all cellular work through oxidative phosphorylation, consuming the oxygen we breathe in the process. They also house the citric acid cycle, beta-oxidation of fatty acids, heme synthesis, steroid hormone biosynthesis, and a major checkpoint of apoptotic cell death (cytochrome c release initiates the intrinsic apoptosis pathway).
The very oxygen that mitochondria require for energy production is also the source of most cellular ROS. During electron transport, approximately 0.2 to 2% of consumed oxygen leaks out of the chain at Complexes I and III, forming superoxide anion (O2·-) instead of being fully reduced to water at Complex IV. In a resting cell at low metabolic rate, that fraction may be only 0.2%. Under metabolic stress, hypoxia-reoxygenation, dysfunction, or genetic compromise of the electron transport chain, the leak can rise to several percent. Multiply by the total oxygen consumed per day (the body uses roughly 350-500 liters of oxygen daily), and the ROS production at the mitochondrial inner membrane is the dominant source of cellular oxidant load.
The same mitochondria are also a primary target of that ROS damage:
- Mitochondrial DNA (mtDNA) sits in the matrix, just nanometers from the electron transport chain leak site, and is not packaged into histones the way nuclear DNA is. It is therefore more vulnerable to ROS damage and accumulates mutations approximately 10x faster than nuclear DNA. Each cell has hundreds to thousands of mtDNA copies (versus 2 nuclear copies), and damaged copies can clonally expand within a cell over time.
- Inner mitochondrial membrane lipids — the cardiolipin that is essential for Complex I, III, IV, V, and the ADP/ATP translocator function — are highly enriched in oxidation-prone polyunsaturated fatty acid chains. Cardiolipin peroxidation directly disrupts respiratory chain assembly and triggers release of cytochrome c into the cytoplasm, initiating apoptosis.
- Iron-sulfur cluster enzymes — the Fe-S clusters in Complex I, Complex II, aconitase (citric acid cycle), and others are exquisitely sensitive to oxidative inactivation.
This sets up the central feedback loop of mitochondrial aging: ROS damages mitochondria, damaged mitochondria leak more ROS, more ROS damages more mitochondria. Cells that cannot break this loop (through mitophagy and biogenesis discussed below) accumulate dysfunctional mitochondria, transition to greater glycolytic dependence, and ultimately senesce or die. This is the modern molecular update on Denham Harman's 1956 free-radical theory of aging and the related Harman 1972 mitochondrial theory of aging.
The Electron Transport Chain and Where the Leak Happens
The mitochondrial electron transport chain is composed of four large protein complexes (Complex I through IV) plus ATP synthase (sometimes called Complex V) embedded in the inner mitochondrial membrane, with two mobile electron carriers (ubiquinone and cytochrome c) shuttling electrons between them. The flow:
- Complex I (NADH dehydrogenase) — receives electrons from NADH (generated by the citric acid cycle and fatty acid oxidation), pumps 4 protons across the inner membrane, passes electrons to ubiquinone. This is the largest leak site for superoxide production, especially under reverse-electron-transport conditions.
- Complex II (succinate dehydrogenase) — receives electrons from succinate (citric acid cycle intermediate via FADH2), passes them to ubiquinone. Does not pump protons. Also a leak site, particularly relevant in reperfusion injury after ischemia.
- Ubiquinone (Coenzyme Q10) — the mobile lipophilic electron carrier moving between Complex I/II and Complex III. Its reduced form (ubiquinol) is itself an antioxidant.
- Complex III (cytochrome bc1 complex) — transfers electrons from ubiquinol to cytochrome c, pumps 4 protons. The Q cycle at Complex III is the second major superoxide leak site.
- Cytochrome c — small water-soluble carrier in the intermembrane space.
- Complex IV (cytochrome c oxidase) — transfers electrons to O2, forming water; pumps 2 protons. This is the actual oxygen consumption step. Iron and copper cofactors.
- ATP Synthase (Complex V) — the proton gradient generated by Complexes I, III, IV is harnessed to drive ATP synthesis from ADP and Pi.
The leak is greatest when:
- The proton gradient is high and ATP demand is low (the chain is "backed up" with reduced cofactors that cannot be re-oxidized fast enough). This is the typical state during caloric excess and sedentary metabolism.
- Specific complexes are inhibited (rotenone at Complex I, antimycin A at Complex III) or damaged.
- CoQ10 is depleted (statin drugs inhibit HMG-CoA reductase, which is also the rate-limiting step for endogenous CoQ10 synthesis).
- The inner membrane lipid composition is compromised (cardiolipin oxidation, deficiency of essential fatty acids).
The leak is reduced by:
- Sufficient ATP demand to maintain electron flow through the chain (exercise).
- Uncoupling proteins (UCP1 in brown adipose, UCP2/3 in other tissues) that allow controlled dissipation of the proton gradient as heat instead of ATP.
- Caloric restriction or intermittent fasting (which transiently reduces substrate flux through the chain).
This last point is mechanistically important. Caloric restriction extends lifespan in nearly every species studied (worms, flies, mice, monkeys), and reduced mitochondrial ROS production from a less-saturated electron transport chain is one of the principal proposed mechanisms.
Coenzyme Q10 (Ubiquinol) — Electron Carrier and Antioxidant
Coenzyme Q10 is the electron carrier shuttling reducing equivalents between Complex I/II and Complex III in the electron transport chain. It is a lipophilic quinone (the 10 in CoQ10 refers to its 10-isoprenoid-unit hydrophobic tail). It exists in two forms: oxidized ubiquinone and reduced ubiquinol. The reduced form is the better-absorbed supplement form and is also the form that has direct antioxidant activity in the membrane.
Endogenous CoQ10 synthesis declines with age (myocardial CoQ10 at age 80 is approximately 50% of age 20 levels) and is suppressed by statin drugs (HMG-CoA reductase is the shared rate-limiting step for both cholesterol synthesis and the polyisoprenoid tail of CoQ10). Statin-induced CoQ10 deficiency may contribute to statin-associated muscle symptoms, though this remains debated and is supported by some meta-analyses (Banach 2015) and refuted by others.
The supplementation evidence:
- Heart failure — the Q-SYMBIO trial (Mortensen 2014) randomized 420 patients with NYHA Class III-IV heart failure to CoQ10 300 mg/day or placebo, added to standard heart failure therapy. After 2 years the CoQ10 group had 43% reduction in major adverse cardiovascular events (death, hospitalization). This is a substantial effect for an OTC supplement.
- Statin-associated myalgia — mixed evidence; some studies positive, others negative. A trial of 100-200 mg/day CoQ10 is reasonable in patients with statin-associated muscle symptoms before discontinuing the statin.
- Migraine prophylaxis — the American Academy of Neurology guideline includes CoQ10 100 mg three times daily as an evidence-based option for migraine prevention.
- Mitochondrial diseases — primary CoQ10 deficiency and other inherited mitochondrial diseases are treated with high-dose CoQ10 (often 1,000-2,400 mg/day).
- Parkinson's disease — the QE3 trial (Beal 2014) of high-dose CoQ10 in early Parkinson's was disappointing for disease modification despite earlier promising pilot data.
- Fertility — CoQ10 200-600 mg/day improves oocyte quality in older women undergoing IVF (Bentov 2014).
Practical dosing: 100-200 mg of ubiquinol daily for general antioxidant and statin support; 200-300 mg/day for heart failure; 300-600 mg/day for migraine or fertility; higher for primary mitochondrial disease under specialist supervision. Ubiquinol is better absorbed than ubiquinone, especially in older adults. Take with a fatty meal for absorption. Brand quality matters — Kaneka Ubiquinol is the most reputable manufacturer.
PQQ and Mitochondrial Biogenesis
Pyrroloquinoline quinone (PQQ) is a small redox-active cofactor found in trace amounts in many foods (parsley, green tea, kiwi, papaya, tofu, natto, breast milk). It was initially studied for its role in bacterial dehydrogenases, then found to be conditionally essential in mammals based on rodent dietary deprivation experiments by Killgore and Stites (2003).
PQQ's major distinct mechanism is induction of mitochondrial biogenesis — the synthesis of new mitochondria from existing ones. It does this by activating the master transcriptional regulator PGC-1alpha (peroxisome proliferator-activated receptor gamma coactivator 1-alpha), which coordinates the transcription of nuclear and mitochondrial genes for mitochondrial proteins. Activated PGC-1alpha increases mitochondrial number and total mitochondrial mass within target tissues.
The human clinical evidence is small but interesting. A 2013 Nakano trial in 17 adults showed PQQ 20 mg/day for 8 weeks reduced fatigue and improved sleep quality. A 2009 Harris trial showed reduced markers of inflammation and oxidative damage. A 2016 trial by Hwang showed improved cognitive function in older adults.
Typical dose: 10-20 mg/day. Often combined with CoQ10 since the two work at complementary stages (CoQ10 supports existing electron transport; PQQ stimulates production of new mitochondria).
NAD+ Decline with Aging and NMN/NR Supplementation
Nicotinamide adenine dinucleotide (NAD+) is the central redox cofactor for cellular metabolism. It accepts electrons from glycolysis, the citric acid cycle, and fatty acid oxidation (becoming NADH), then donates them to the electron transport chain (regenerating NAD+). The total NAD+ + NADH pool is therefore continuously recycled, but the size of that total pool also matters for several other functions: NAD+ is a substrate for the sirtuin family (especially SIRT1 and SIRT3), for PARP enzymes (DNA damage repair), and for cyclic ADP-ribose generation (calcium signaling).
Cellular NAD+ declines with aging — by approximately 50% from young adulthood to old age in tissues studied. The decline is driven by increased degradation (the CD38 enzyme, induced by chronic inflammation, consumes NAD+) and reduced synthesis. NAD+ decline limits sirtuin activity, impairs DNA damage repair, reduces oxidative phosphorylation efficiency, and is associated with several age-related changes.
Two major NAD+ precursors are now available as supplements:
- Nicotinamide riboside (NR) — sold as Niagen and similar trademarks. Multiple human trials (Trammell 2016, Martens 2018, Conze 2019) have shown 30-100% increases in blood NAD+ with 250-1,000 mg/day for several weeks. Clinical endpoints have been more modest — some metabolic improvements, blood pressure reduction in older adults.
- Nicotinamide mononucleotide (NMN) — David Sinclair's preferred precursor. Multiple recent human trials (Yoshino 2021, Liao 2021) have shown similar NAD+ elevation. Direct head-to-head comparisons with NR are limited; either works for raising blood NAD+.
The remaining open questions: does raising blood NAD+ actually translate to meaningful clinical benefits in healthy aging? Does it slow age-related disease? Sinclair's mouse work was striking, but human translation has been measured rather than dramatic. The supplements are well-tolerated and reasonable as part of a longevity stack; the cost-benefit at this stage of the evidence is modest.
Typical dose: NR 300-1,000 mg/day or NMN 250-1,000 mg/day. Take in the morning (some users report sleep disruption from evening doses). Pairs well with resveratrol or pterostilbene (sirtuin activators).
Alpha-Lipoic Acid as the Mitochondrial Cofactor
Alpha-lipoic acid (ALA, thioctic acid) is both an essential mitochondrial enzyme cofactor and an antioxidant supplement in its own right. As a cofactor it is bound to the lysine residue of three multienzyme complexes: pyruvate dehydrogenase (links glycolysis to citric acid cycle), alpha-ketoglutarate dehydrogenase (citric acid cycle), and branched-chain alpha-keto acid dehydrogenase (branched-chain amino acid catabolism). Without lipoic acid these enzymes cannot function.
As a supplement, alpha-lipoic acid has unique properties: it is both water-soluble and lipid-soluble (it dissolves in both compartments, unlike Vitamin C which is water-only and Vitamin E which is lipid-only); it is reduced inside cells to dihydrolipoic acid, which is itself a potent antioxidant; it regenerates other antioxidants (Vitamins C and E, glutathione); it chelates redox-active metal ions (iron, copper) that catalyze Fenton chemistry; and it crosses the blood-brain barrier.
The strongest clinical evidence is for diabetic peripheral neuropathy. The SYDNEY 2 trial (Ametov 2003) and ALADIN trials randomized over 600 patients to alpha-lipoic acid 600-1,800 mg/day or placebo for 5-19 weeks. The ALA groups had significant reductions in neuropathic pain scores. The European Federation of Neurological Societies recommends alpha-lipoic acid as evidence-based therapy for diabetic neuropathy. Typical regimen: 600 mg/day oral; IV protocols use 600 mg/day for 3 weeks in severe cases.
Other applications with reasonable evidence:
- Insulin sensitivity — modest improvements in HOMA-IR in metabolic syndrome.
- Heavy metal chelation adjunct — supports the body's handling of mercury, lead, cadmium (not a substitute for DMSA in true heavy metal toxicity).
- Mitochondrial dysfunction conditions — often included in mitochondrial-support stacks.
- Liver protection — historically used (under brand name Thioctacid) for amanita mushroom poisoning and chronic hepatitis.
Typical dose: 300-600 mg/day, taken on an empty stomach for better absorption (food reduces bioavailability). The R-isomer is the natural and more bioactive form (most supplements are racemic R/S); R-alpha-lipoic acid 300 mg/day is approximately equivalent to racemic 600 mg/day. Caution in diabetics on insulin or sulfonylureas: alpha-lipoic acid can lower blood glucose and may require medication adjustment.
Exercise — The Most Potent Mitochondrial Biogenesis Stimulus
No supplement — not CoQ10, PQQ, NMN, NR, or any combination — comes close to exercise as a stimulus for mitochondrial biogenesis. Endurance exercise activates the AMPK and PGC-1alpha signaling pathways far more potently than any pharmacologic agent. Trained endurance athletes have approximately 2-3x the mitochondrial density in skeletal muscle compared to sedentary adults of the same age. The mitochondrial machinery of an active 70-year-old can match or exceed that of a sedentary 30-year-old.
The relevant mechanisms:
- AMP/ATP ratio rises during exercise — ATP demand exceeds production, ADP and AMP accumulate, activating AMPK (AMP-activated protein kinase). AMPK in turn activates PGC-1alpha, which drives mitochondrial biogenesis.
- Calcium oscillations — muscle contraction releases calcium from sarcoplasmic reticulum, activating calcium-calmodulin-dependent protein kinases that also signal to PGC-1alpha.
- Transient ROS spikes — exercise-induced ROS at moderate concentrations is the hormetic signal that triggers adaptive antioxidant responses — including Nrf2 activation, glutathione synthesis upregulation, and antioxidant enzyme induction. This is why supplementing with megadose antioxidants during exercise training can blunt the adaptive benefit (Ristow 2009 PNAS demonstrated that Vitamin C and E supplements abolished the insulin-sensitizing effect of exercise training).
- Substrate cycling — alternating fuel sources during prolonged exercise (glycogen, fatty acids, ketones) trains the metabolic flexibility of mitochondria.
The exercise prescription with the strongest evidence for mitochondrial benefit:
- Aerobic base training — 150-300 minutes per week of moderate-intensity aerobic exercise (walking, cycling, swimming) at a level where you can hold a conversation. This drives the bulk of mitochondrial biogenesis.
- High-intensity interval training (HIIT) — 2-3 sessions per week of short intervals near maximal effort (Tabata protocol, 4x4 protocol). HIIT produces more mitochondrial adaptation per unit of training time than steady-state training, especially relevant when time is limited.
- Resistance training — 2-3 sessions per week to maintain muscle mass (mitochondria-rich tissue) and prevent the sarcopenia that contributes to mitochondrial decline.
- Avoid antioxidant megadosing during the training period — if optimizing exercise adaptation, take antioxidant supplements at times of day separated from training, or skip them on training days.
This is the most important intervention on this page. Supplements have a useful role; exercise is foundational.
Mitophagy: Removing Damaged Mitochondria
Mitochondrial quality control depends not just on producing new mitochondria but on removing damaged ones. The selective autophagy of mitochondria is called mitophagy. The two best-characterized pathways:
- PINK1/Parkin pathway — under normal conditions, the kinase PINK1 is imported into healthy mitochondria and proteolytically degraded. When a mitochondrion loses its membrane potential (a marker of damage), PINK1 import fails, PINK1 accumulates on the outer mitochondrial membrane, phosphorylates ubiquitin, and recruits the E3 ubiquitin ligase Parkin. Parkin then poly-ubiquitinates outer membrane proteins, tagging the damaged mitochondrion for autophagosomal engulfment and lysosomal degradation. Mutations in PINK1 or PARK2/Parkin cause autosomal-recessive early-onset Parkinson's disease — reflecting the importance of mitochondrial quality control specifically in dopaminergic neurons.
- BNIP3 and BNIP3L/Nix pathway — receptor-mediated mitophagy through specific outer membrane receptors that interact directly with the autophagy machinery (LC3-binding motifs).
Mitophagy declines with aging, contributing to the accumulation of dysfunctional mitochondria. Interventions that enhance mitophagy:
- Caloric restriction and intermittent fasting — activate AMPK and inhibit mTOR, both of which promote autophagy and mitophagy.
- Exercise — transiently elevates AMPK, promoting mitophagy alongside biogenesis.
- Urolithin A — the gut-microbiota metabolite of ellagitannins (from pomegranate, walnuts, berries) has been shown in mice and humans to enhance mitophagy. The Mitopure supplement provides 500 mg urolithin A directly.
- Spermidine — found in wheat germ, soybeans, mushrooms; enhances autophagy across species. Several pilot human trials suggest cognitive and metabolic benefits.
- Rapamycin — mTOR inhibitor; off-label longevity use under specialist supervision.
The Andreux et al. 2019 trial in healthy older adults showed urolithin A 500-1,000 mg/day for 28 days induced mitochondrial gene expression and improved markers of mitochondrial function. The longer-term Singh 2022 trial showed improvements in muscle endurance.
Mitochondrial Dysfunction as Unifying Disease Mechanism
Mitochondrial dysfunction is now recognized as a contributing factor in a broad range of chronic diseases — not necessarily the primary cause, but a converging downstream mechanism that amplifies pathology and creates targets for intervention.
- Type 2 diabetes and insulin resistance — mitochondrial overload from chronic caloric excess and the resulting incomplete fatty acid oxidation generates lipid intermediates (diacylglycerol, ceramide) that disrupt insulin signaling. Mitochondrial biogenesis defects (PGC-1alpha downregulation) are characteristic of insulin-resistant skeletal muscle.
- Cardiovascular disease — the heart consumes more energy per gram than any other tissue and is exquisitely mitochondrial-dependent. Heart failure features reduced mitochondrial efficiency. The Q-SYMBIO trial result with CoQ10 reflects this.
- Neurodegeneration — Parkinson's disease has specific Complex I deficits in substantia nigra. Alzheimer's features mitochondrial dysfunction preceding amyloid deposition. ALS has mitochondrial calcium handling defects. Huntington's disease has Complex II inhibition.
- Chronic fatigue syndrome / ME-CFS — the Naviaux 2016 metabolomics work showed a coordinated "dauer-like" hypometabolic state with multiple mitochondrial impairments.
- Fibromyalgia — reduced muscle mitochondrial CoQ10 and Complex I activity.
- Sarcopenia and frailty — loss of muscle mitochondrial density is a major contributor to age-related strength and endurance decline.
- Cancer — the Warburg effect (aerobic glycolysis) reflects mitochondrial reprogramming in cancer cells; mitochondrial dysfunction may be both consequence and contributor.
- Aging itself — the mitochondrial theory of aging proposes that accumulated mitochondrial damage is a central driver of the aging phenotype. The evidence is incomplete — the mtDNA mutator mouse develops accelerated aging, but lifelong antioxidant supplementation in many studies does not extend lifespan as cleanly as predicted.
The therapeutic implication is that mitochondrial support is a reasonable adjunct in many chronic disease contexts. Not as primary therapy for any specific condition, but as supportive therapy that improves underlying cellular bioenergetics.
Cautions and Practical Stack
- CoQ10 and blood thinners — CoQ10 has mild structural similarity to vitamin K and theoretically can reduce the effect of warfarin; monitor INR more closely when starting or stopping CoQ10. No interaction with direct oral anticoagulants (apixaban, rivaroxaban).
- Alpha-lipoic acid and diabetes drugs — ALA lowers blood glucose; insulin and sulfonylurea doses may need adjustment.
- NMN/NR and cancer — theoretical concern that boosting NAD+ might support cancer cell metabolism; avoid in patients with active known malignancy until oncologist consulted.
- Resveratrol and bleeding — mild antiplatelet effect; caution combining with anticoagulants.
- Antioxidant timing around exercise — per Ristow 2009 PNAS, megadose Vitamin C and E during exercise training blunts insulin-sensitization benefit. Separate antioxidant supplements from training by several hours or skip on training days.
- Methylene blue — emerging mitochondrial-support agent; potent monoamine oxidase inhibitor; do not combine with SSRI, SNRI, MAOI, or tramadol due to serotonin syndrome risk.
- Mitochondrial poisons — rotenone (pesticide), antimycin A (research chemical), cyanide, carbon monoxide, oligomycin, amobarbital, certain HIV drugs (zidovudine, didanosine, stavudine). Awareness rather than action item.
- Statin-CoQ10 interaction — if a patient on statin therapy develops muscle symptoms, a trial of CoQ10 100-200 mg/day for 4-6 weeks before discontinuing the statin is reasonable. Some patients improve dramatically; others do not.
A reasonable mitochondrial-support stack for a healthy adult over 50 prioritizing healthspan:
- Aerobic exercise 150-300 min/week + 2 HIIT sessions + 2-3 resistance sessions
- Mediterranean-pattern diet, ample polyphenols, time-restricted eating window (12-16 hours)
- Ubiquinol 100-200 mg/day (more if on statin or with heart disease)
- Alpha-lipoic acid 300-600 mg/day on empty stomach
- NMN or NR 250-500 mg/day in the morning
- PQQ 10-20 mg/day (optional, paired with CoQ10)
- Urolithin A 500 mg/day or 1 ounce walnuts plus pomegranate seeds daily
- Magnesium 200-400 mg/day (broad mitochondrial enzyme cofactor)
- B-complex with methylated forms
This is one rational stack; many variations are equally defensible. The exercise and diet portions are non-negotiable; the supplements are useful adjuncts.
Key Research Papers
- Harman D (1972). The biologic clock: the mitochondria? Journal of the American Geriatrics Society. — PubMed
- Mortensen SA et al. (2014). The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO. JACC Heart Failure. — PubMed
- Banach M et al. (2015). Statin therapy and plasma coenzyme Q10 concentrations — a systematic review and meta-analysis. Pharmacological Research. — PubMed
- Trammell SAJ et al. (2016). Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nature Communications. — PubMed
- Martens CR et al. (2018). Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nature Communications. — PubMed
- Yoshino M et al. (2021). Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science. — PubMed
- Andreux PA et al. (2019). The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nature Metabolism. — PubMed
- Ristow M et al. (2009). Antioxidants prevent health-promoting effects of physical exercise in humans. PNAS. — PubMed
- Ametov AS et al. (2003). The sensory symptoms of diabetic polyneuropathy are improved with alpha-lipoic acid (SYDNEY trial). Diabetes Care. — PubMed
- Bentov Y et al. (2014). Coenzyme Q10 supplementation and oocyte aneuploidy in women undergoing IVF-ICSI treatment. Clinical Medicine Insights: Reproductive Health. — PubMed
- Wallace DC (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer. Annual Review of Genetics. — PubMed
- Naviaux RK et al. (2016). Metabolic features of chronic fatigue syndrome. PNAS. — PubMed
PubMed Topic Searches
- PubMed: Mitochondrial ROS production
- PubMed: CoQ10 clinical trials
- PubMed: NMN/NR human trials
- PubMed: Mitophagy and urolithin
- PubMed: Exercise mitochondrial biogenesis
Connections
- Oxidative Stress Hub
- Oxidative Stress Benefits Hub
- Glutathione Master Antioxidant
- NAC and Precursors
- Polyphenol Foods
- Coenzyme Q10
- NAD+ (NMN/NR)
- Alpha-Lipoic Acid
- Magnesium
- Heart Failure
- Parkinson's Disease
- Type 2 Diabetes
- Exercise
- Intermittent Fasting
- Mediterranean Diet