Morley Robbins on Copper-Iron Imbalance
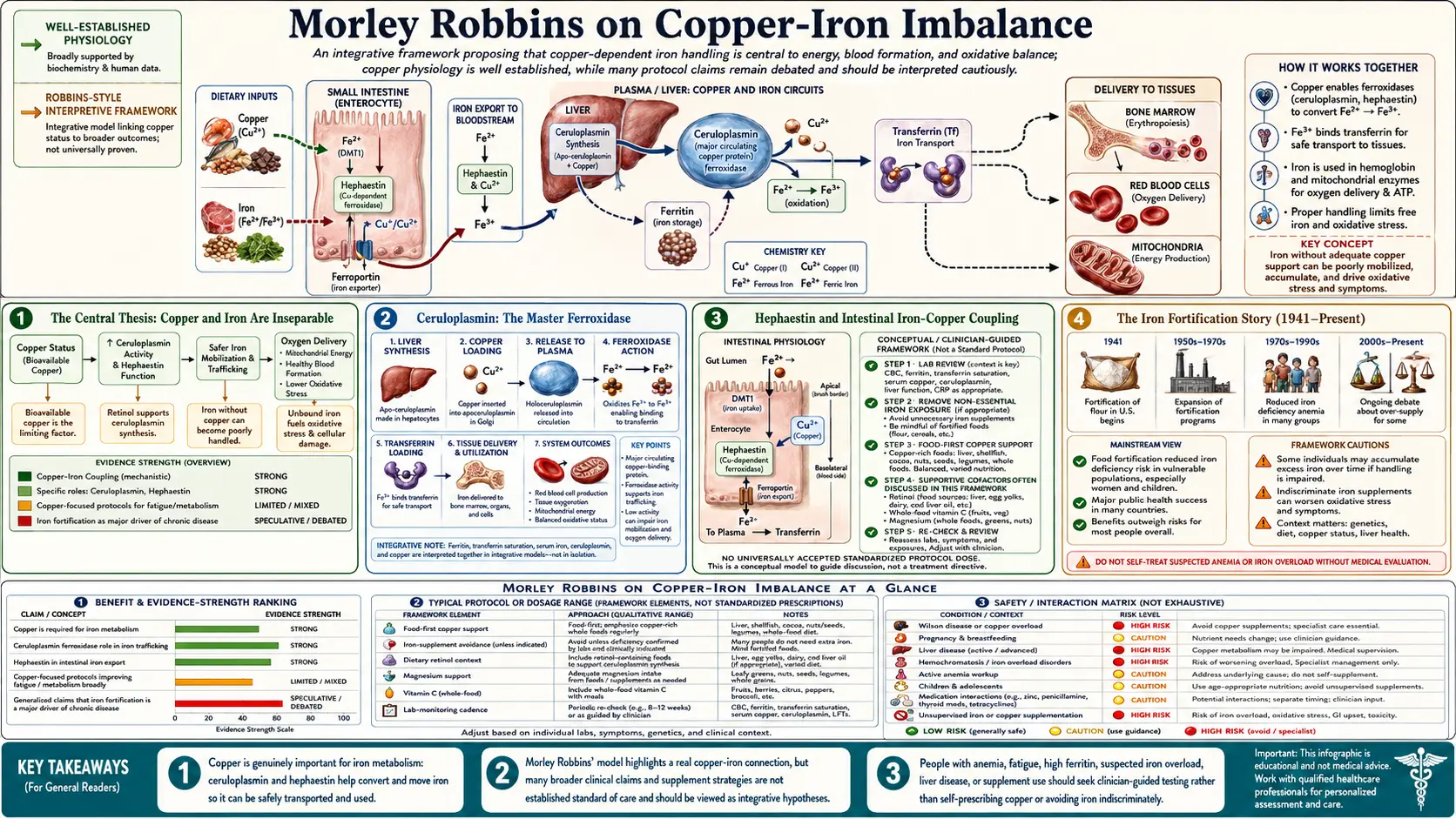
Morley Robbins' central physiological claim is that copper and iron are not independent minerals — they are obligate partners in a single redox-coupled system, and a deficiency of bioavailable copper inevitably leads to tissue iron overload, oxidative stress, ferroptosis, and the symptom complex (fatigue, anxiety, hair loss, insomnia, hypothyroid features, autoimmune flares) that drives most chronic-illness office visits. The biochemistry is well-established at the bench: ceruloplasmin, the copper-dependent ferroxidase, is required to oxidize ferrous iron (Fe-2+) to ferric iron (Fe-3+) so it can be loaded onto transferrin and safely transported. Without functional ceruloplasmin, iron accumulates as unbound "free iron" in tissues, where it catalyzes Fenton-reaction hydroxyl radical formation and drives the lipid peroxidation cascade that ends in ferroptotic cell death. This page walks through the biochemistry, the historical context of iron fortification, the lab markers that reveal the imbalance, and Robbins' therapeutic protocol.
Table of Contents
- The Central Thesis: Copper and Iron Are Inseparable
- Ceruloplasmin: The Master Ferroxidase
- Hephaestin and Intestinal Iron-Copper Coupling
- The Iron Fortification Story (1941-Present)
- Why Serum Ferritin Is Misread by Conventional Medicine
- Ferroptosis: How Excess Iron Kills Cells
- The Clinical Syndrome of Functional Copper Deficiency
- The Robbins Therapeutic Protocol
- Cautions and Contraindications
- Key Research Papers
- Connections
- Featured Videos
The Central Thesis: Copper and Iron Are Inseparable
Conventional medicine treats copper and iron as two independent minerals on the periodic table. Robbins' argument, drawing on the work of Earl Frieden, Maria Linder, and Leland Klevay, is that this is biochemically incorrect. In every multicellular organism studied, iron metabolism is enzymatically coupled to copper metabolism through a small set of copper-dependent oxidases that are absolutely required for iron to move safely from the gut into the bloodstream and from the bloodstream into the bone marrow for hemoglobin synthesis.
The two key enzymes are ceruloplasmin (a serum protein synthesized in the liver that requires six copper atoms per molecule to be enzymatically active) and hephaestin (the membrane-bound copper-dependent ferroxidase expressed on the basolateral surface of intestinal enterocytes). Together they perform the same chemistry — converting ferrous iron (Fe-2+) to ferric iron (Fe-3+) — at two different anatomical sites. Without them, iron cannot be loaded onto its carrier protein transferrin, cannot move across cell membranes safely, and accumulates in the labile iron pool where it catalyzes oxidative damage.
The consequence: when bioavailable copper falls, iron does not necessarily fall too. Total body iron may stay constant or even rise (because hepcidin-mediated efflux from cells requires the same ferroxidase chemistry), but the iron is in the wrong place — deposited in tissues (liver, heart, brain, joints, endocrine organs) rather than circulating safely on transferrin. This is what Robbins calls "iron dysregulation," and it is the foundation of his clinical model.
Ceruloplasmin: The Master Ferroxidase
Ceruloplasmin is an alpha-2 glycoprotein synthesized in hepatocytes and secreted into the bloodstream. Its primary enzymatic function is ferroxidation — the oxidation of Fe-2+ to Fe-3+ so the iron can bind to transferrin. Each ceruloplasmin molecule contains six tightly bound copper atoms; without all six copper atoms loaded, the protein still circulates and can be measured by immunoassay, but it lacks enzymatic activity. This is the critical distinction between immunoreactive ceruloplasmin (what most labs measure) and enzymatic ceruloplasmin (what actually performs the biological function).
Robbins emphasizes that a "normal" serum ceruloplasmin of 25-35 mg/dL on a routine lab does not guarantee functional ferroxidase activity. The protein may be there but unloaded with copper. The way to confirm functional activity is to measure the enzymatic (oxidase) ceruloplasmin specifically — a less common assay typically available through specialty reference labs — or to back-calculate from the relationship between serum copper and ceruloplasmin (each mg/dL of ceruloplasmin should bind roughly 0.03 micrograms of copper; substantial deviation suggests under-loading).
The functional ceruloplasmin assay also addresses one of the persistent confusions in functional medicine: patients with serum copper that is elevated above the reference range are sometimes told they have "copper toxicity" and should reduce copper intake. Robbins argues this is often backwards — the elevated serum copper is "free" or non-ceruloplasmin-bound copper that has nowhere to go because ceruloplasmin is unloaded. The correct intervention is to support ceruloplasmin synthesis (retinol, magnesium, whole-food copper) so the existing copper can be properly bound and put to work.
Hephaestin and Intestinal Iron-Copper Coupling
Hephaestin is the lesser-known but equally important sibling of ceruloplasmin. It is a membrane-bound copper-dependent ferroxidase expressed on the basolateral (blood-facing) side of intestinal enterocytes, particularly in the proximal small intestine. Its function is to oxidize Fe-2+ to Fe-3+ at the moment iron exits the enterocyte through the ferroportin transporter, so the freshly-absorbed iron can immediately bind to transferrin in the portal venous system.
Hephaestin was first characterized in the late 1990s as the gene mutated in the sex-linked anemia (sla) mouse strain. The sla mouse has normal iron intake, normal duodenal iron absorption into the enterocyte, but profoundly impaired iron export from the enterocyte to the bloodstream because the copper-dependent ferroxidase step fails. The result is iron trapped in enterocytes, which die and slough off, and systemic iron deficiency despite adequate dietary intake.
The human implications are substantial. Patients with chronic dietary copper insufficiency, with celiac disease that has destroyed the enterocyte brush border, with bariatric surgery that has bypassed the duodenum, or with chronic zinc supplementation that displaces copper, can have impaired hephaestin function and therefore impaired iron absorption. The conventional response — iron supplementation — is futile until the copper-dependent ferroxidase machinery is restored. This is one of the mechanistic explanations for the common clinical observation that iron deficiency anemia persists despite years of oral iron supplementation.
The Iron Fortification Story (1941-Present)
The wartime War Food Administration mandated iron fortification of refined wheat flour and breakfast cereals in the United States in 1941, initially as a public health response to the discovery that conscripts from low-income populations had high rates of frank iron deficiency anemia. The fortification standard required adding electrolytic iron, ferrous fumarate, or ferrous sulfate to enriched flour at 20 mg per pound (and later levels). The intervention was, at the time, a defensible public health choice given the iron deficiency rates documented in Sherman's and Sebrell's field surveys of the 1930s.
The consequence eight decades later is that almost every grain product in the conventional American food supply — bread, cereal, pasta, baked goods — is fortified with synthetic inorganic iron. A typical American adult consumes 15-20 mg/day of iron, of which a substantial fraction comes from fortified grain. Combined with iron-fortified infant formula and the historical clinical practice of recommending iron supplements for any patient with fatigue or borderline-low ferritin, this has produced a population in which a meaningful fraction of adults — particularly post-menopausal women and men — have iron excess rather than iron deficiency.
Robbins points to the NHANES data showing serum ferritin values that have risen substantially over the past 50 years in U.S. adults, and to the rising prevalence of hemochromatosis-spectrum disease, fatty liver, and iron-driven cardiovascular and neurodegenerative pathology, as evidence that the cumulative effect of iron fortification has crossed a population threshold from net benefit to net harm in a substantial subset of adults. He advocates for ferritin targets of 30-50 ng/mL in women and 50-70 ng/mL in men, with anything sustained above 80 ng/mL flagged for evaluation — substantially below the conventional reference range upper limits of 200-400 ng/mL.
Why Serum Ferritin Is Misread by Conventional Medicine
Serum ferritin is one of the most consequentially-misread lab markers in modern medicine. Conventional teaching treats serum ferritin as a clean proxy for total body iron stores, and the conventional reference range (typically 30-400 ng/mL in men, 15-200 ng/mL in women) reflects what is statistically common in a general population, not what is biologically optimal.
Two problems with this framing:
- Ferritin is an acute-phase reactant. Inflammation of any kind — infection, autoimmunity, obesity-associated inflammation, recent vaccination, even significant exercise — elevates serum ferritin independently of iron stores. A "normal" or elevated ferritin in an inflamed patient may coexist with frank iron deficiency in the bone marrow. The relevant assessment requires also measuring CRP or another inflammatory marker and interpreting ferritin in context.
- The upper reference range is statistically common, not biologically optimal. The 95th percentile of an iron-fortified population includes a substantial fraction of subjects with subclinical iron overload, fatty liver, and metabolic syndrome. Treating the 95th percentile as "normal" embeds the population pathology into the reference range.
Robbins' preferred ferritin targets — 30-50 ng/mL women, 50-70 ng/mL men — sit at the lower end of the conventional reference range and align with the Iron Disorders Institute recommendations and with the hemochromatosis-prevention literature. The argument is mechanistic, not arbitrary: above approximately 70 ng/mL in men, evidence of subclinical hepatic iron deposition begins to appear, and above approximately 200 ng/mL the risk of metabolic and cardiovascular pathology begins to rise meaningfully.
Ferroptosis: How Excess Iron Kills Cells
The mechanistic linkage between iron excess and tissue damage is now well-characterized through the ferroptosis literature, a research area that exploded after Dixon and Stockwell coined the term in 2012. Ferroptosis is an iron-dependent form of regulated cell death distinct from apoptosis, necrosis, and autophagy. It is triggered when iron-catalyzed lipid peroxidation overwhelms the cellular antioxidant defense (particularly glutathione peroxidase 4, GPX4), producing lipid hydroperoxides that destroy membrane integrity and kill the cell.
The biochemistry sits squarely in the Fenton reaction: Fe-2+ + H2O2 yields Fe-3+ + OH- + the hydroxyl radical (.OH), the most reactive oxygen species in biology. The hydroxyl radical attacks the polyunsaturated fatty acid (PUFA) chains in cell membranes, abstracting hydrogen atoms and initiating the lipid peroxidation cascade. Healthy cells contain enough GPX4 to neutralize the propagating peroxyl radicals; cells with excess labile iron and depleted GPX4 cannot keep up, and the cascade runs to ferroptotic death.
Ferroptosis is now implicated in a broad list of clinical conditions: hepatocellular carcinoma, ischemia-reperfusion injury, neurodegenerative disease (Alzheimer's, Parkinson's, Huntington's), kidney disease, and several cancers. Pharma is actively developing ferroptosis-inducing drugs for cancer (the same chemistry that kills neurons in Alzheimer's can be turned against cancer cells). Robbins' framework predates the ferroptosis literature but predicted its essential conclusion: that excess iron in tissues drives oxidative cell death, and that copper-dependent ferroxidases are the primary biological defense.
The Clinical Syndrome of Functional Copper Deficiency
The clinical syndrome Robbins associates with functional copper deficiency / iron dysregulation is recognizable to any functional medicine practitioner who sees the same patient population over time. The features include:
- Persistent fatigue not responsive to thyroid, adrenal, or B-vitamin support — explained mechanistically by impaired mitochondrial cytochrome c oxidase (the copper-dependent terminal electron-transport complex)
- Cold intolerance and hypothyroid symptoms with sometimes-normal TSH — explained by copper-dependent T3 conversion
- Hair loss, premature graying, and connective tissue laxity — explained by lysyl oxidase and tyrosinase (both copper-dependent)
- Anxiety and insomnia — partly magnesium, partly the catecholamine effects of dopamine-beta-hydroxylase (copper-dependent)
- Recurrent infections — phagocyte superoxide dismutase (SOD1, SOD3) is copper-dependent; impaired innate immunity is one consequence
- Hyperferritinemia with normal or low hemoglobin — the "anemia of chronic disease" pattern, often present
- Elevated liver enzymes (ALT, AST) and fatty liver imaging — the hepatic iron deposition that accompanies long-standing imbalance
- Failure to respond to iron supplementation — oral iron is poorly absorbed and poorly utilized in the absence of functional copper enzymes
The differential diagnosis includes hereditary hemochromatosis, transfusion-dependent thalassemia, chronic liver disease of other cause, and Wilson's disease (the rare opposite syndrome of copper toxicity). The RCP Lab Panel is designed to differentiate these and identify the specific subtype of copper-iron dysregulation present. See our RCP Lab Panel page for the full workup.
The Robbins Therapeutic Protocol
The therapeutic approach in the Root Cause Protocol is built around "Stops" (things to stop doing) and "Starts" (things to start doing). The Stops list and Starts list are detailed in Cure Your Fatigue and on the official Root Cause Protocol website; the abbreviated copper-iron balance version is:
Stops:
- Stop all synthetic iron supplements unless documented frank iron deficiency anemia with low transferrin saturation
- Stop iron-fortified foods where practical (most refined-grain products)
- Stop ascorbic acid (synthetic isolated vitamin C) supplements above 250 mg/day — high-dose AA at this exposure level is argued to displace copper from ceruloplasmin and increase intestinal iron absorption
- Stop high-dose isolated zinc supplements (above ~15-25 mg/day) without simultaneous copper, as zinc competitively inhibits intestinal copper absorption
- Stop synthetic vitamin D supplements at high doses without simultaneous magnesium (synthetic D activation depletes magnesium)
- Stop calcium supplements except as part of a balanced mineral-replete food matrix
Starts:
- Start whole-food copper sources: beef liver 1-2 oz daily, oysters, raw cacao, bee pollen (see Whole Food Copper Sources)
- Start magnesium glycinate or other well-tolerated form, titrated to RBC magnesium target (see Magnesium Foundation)
- Start whole-food vitamin C sources (camu camu, acerola, food-form supplements) rather than isolated ascorbic acid
- Start whole-food vitamin A from cod liver oil or beef liver for ceruloplasmin support
- Start sun exposure as the preferred vitamin D source
- Start unrefined sea salt rather than refined sodium chloride
- For iron-overloaded patients, start regular blood donation (typically every 8-12 weeks for men and post-menopausal women) as the most direct way to reduce body iron burden
The full protocol involves a longer Stops/Starts list and individualization based on the RCP lab panel results. Robbins emphasizes that the protocol is not a treatment for any specific disease — it is a foundational mineral support that allows the body to self-correct downstream symptoms.
Cautions and Contraindications
- Pregnancy: do not reduce iron supplementation or attempt iron reduction in pregnancy without obstetric guidance. Pregnancy expands plasma volume by 40-50% and requires significant iron for fetal hemoglobin synthesis. The RCP approach is generally not adopted during active pregnancy.
- Menstruating women with documented iron deficiency anemia: if transferrin saturation is genuinely low (<15%) and ferritin is genuinely low (<30 ng/mL), this is iron deficiency anemia, not iron dysregulation, and warrants conventional iron supplementation alongside copper-supportive measures.
- Hereditary hemochromatosis: patients with HFE genetic variants require formal hematology evaluation and ongoing therapeutic phlebotomy management. The RCP framework can complement but does not replace standard hemochromatosis care.
- Wilson's disease: the opposite syndrome of copper toxicity from ATP7B gene mutation. Patients with elevated unbound copper and low ceruloplasmin from Wilson's require chelation therapy (penicillamine, trientine) and dietary copper restriction — the opposite of the RCP approach. Genetic testing is recommended before pursuing copper-loading interventions in any patient with hepatic or neuropsychiatric symptoms of unclear etiology.
- Anemia of chronic kidney disease: impaired EPO production and uremic interference with iron utilization create a complex iron picture that often requires specialist management.
- Blood donation for iron reduction: requires hemoglobin above the donor center cutoff (typically 12.5 g/dL women, 13.0 g/dL men). Do not pursue donation if hemoglobin is below this threshold.
Key Research Papers
- Vashchenko G, MacGillivray RT (2013). Multi-copper oxidases and human iron metabolism. Nutrients. — PubMed
- Vulpe CD et al. (1999). Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. — PubMed
- Hellman NE, Gitlin JD (2002). Ceruloplasmin metabolism and function. Annu Rev Nutr. — PubMed
- Harris ZL et al. (1995). Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. PNAS. — PubMed
- Dixon SJ et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. — PubMed
- Stockwell BR et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. — PubMed
- Klevay LM (2000). Cardiovascular disease from copper deficiency — a history. J Nutr. — PubMed
- Sullivan JL (1981). Iron and the sex difference in heart disease risk. Lancet. — PubMed
- Frieden E (1986). Perspectives on copper biochemistry. Clin Physiol Biochem. — PubMed
- Linder MC, Hazegh-Azam M (1996). Copper biochemistry and molecular biology. Am J Clin Nutr. — PubMed
- Salonen JT et al. (1992). High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation. — PubMed
- Pietrangelo A (2010). Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. — PubMed
PubMed Topic Searches
- PubMed: Ceruloplasmin and iron homeostasis
- PubMed: Copper deficiency and iron absorption
- PubMed: Ferroptosis and disease
- PubMed: Ferritin interpretation
- PubMed: Iron fortification and overload
Connections
- Morley Robbins Hub
- Benefits Deep Dive
- Whole Food Copper Sources
- Magnesium Foundation
- RCP Lab Panel
- Iron Overload Hidden Toxicity
- Ceruloplasmin & Bioavailable Copper
- Copper-Iron Dysregulation
- Root Cause Protocol
- Copper
- Iron
- Magnesium
- Organ Meats (Beef Liver)
- Lab Tests
- All Remedies