Cryptogenic Organizing Pneumonia
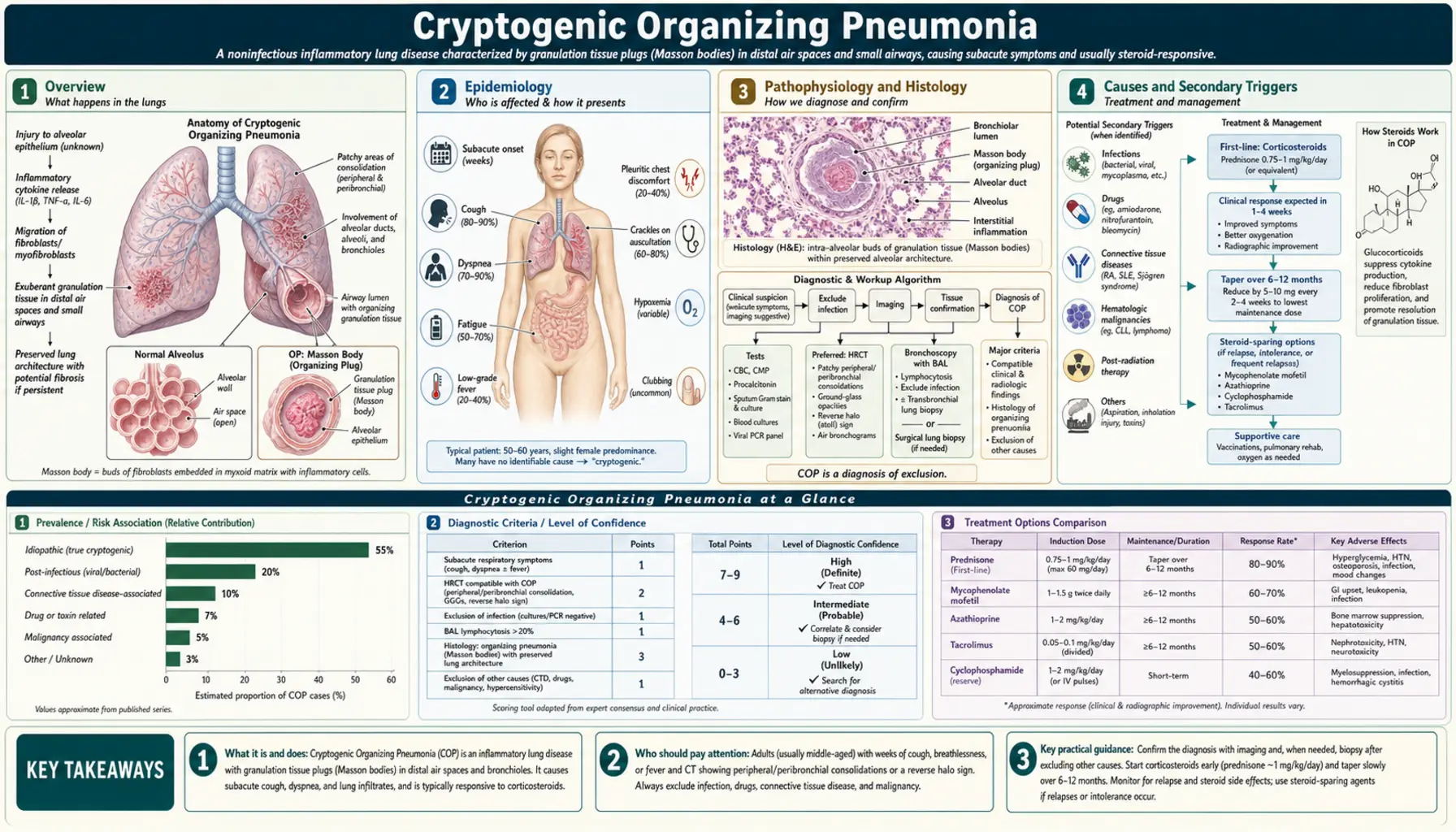
- Overview
- Epidemiology
- Pathophysiology and Histology
- Causes and Secondary Triggers
- Clinical Presentation
- Imaging Findings
- Diagnosis
- Treatment
- Prognosis and Recurrence
- Recent Research
- Research Papers
- Connections
- Featured Videos
1. Overview
Cryptogenic organizing pneumonia (COP) is a distinctive clinicopathological syndrome defined by a specific histological pattern — organizing pneumonia (OP) — in the absence of an identifiable cause. The term "cryptogenic" signals idiopathic origin; the same histological pattern arising from an identifiable cause is called "secondary organizing pneumonia." COP was originally termed bronchiolitis obliterans organizing pneumonia (BOOP) — coined by Epler and colleagues and by King and colleagues in 1985 — but this name was abandoned because the word "bronchiolitis obliterans" implies fixed airway obstruction (as in constrictive bronchiolitis), when in fact COP causes restriction, not obstruction, and is reversible.
COP is part of the idiopathic interstitial pneumonias (IIPs) classification of the American Thoracic Society and European Respiratory Society (2013 update), where it holds a favorable position as the most steroid-responsive and best-prognosis IIP. The hallmark is Masson bodies — intraluminal buds of granulation tissue (fibroblasts embedded in myxoid stroma) filling alveolar ducts and alveoli — which resolve completely with corticosteroids without leaving fibrosis.
2. Epidemiology
COP is a rare condition. Estimated incidence is 1–3 cases per 100,000 per year in population-based studies, and it is likely underdiagnosed. COP affects adults predominantly in the fifth and sixth decades of life (mean age 55–60 at diagnosis), without strong sex predominance, though some series report a slight female predominance. Smokers are not disproportionately affected (unlike many other interstitial lung diseases) — COP occurs in both smokers and non-smokers.
There is no known geographic, occupational, or genetic predisposition for the cryptogenic form. Secondary organizing pneumonia may follow specific exposures (see Section 4). Population data improved after the 2002 ATS/ERS reclassification renamed BOOP to COP, clarifying the epidemiological picture by establishing consistent diagnostic terminology across centers worldwide.
3. Pathophysiology and Histology
The pathological hallmark of COP is the organizing pneumonia (OP) pattern — specific and reproducible on surgical lung biopsy. Understanding the histology is essential because the histological pattern determines both diagnosis and treatment choice.
Masson Bodies
Plugs of granulation tissue composed of young fibroblasts, myxomatous connective tissue, and inflammatory cells fill alveolar ducts, alveoli, and sometimes terminal bronchioles. These are buds — not obliterating plugs that scar — and when they resolve under corticosteroid treatment, lung architecture returns to normal. This reversibility is the critical distinction from usual interstitial pneumonia (UIP/IPF) and from constrictive bronchiolitis.
Preserved Alveolar Architecture
The alveolar walls remain intact with only mild lymphocytic infiltrate. There is no significant honeycomb change or fibroblastic foci, which are defining features of UIP/IPF. Masson bodies are distributed in a patchy, temporally uniform fashion — all lesions are at the same stage of development, unlike the temporal heterogeneity that characterizes UIP.
Inflammatory Infiltrate
Alveolar walls show mild lymphocyte and plasma cell infiltration. Intra-alveolar macrophages and foamy cells are present. Eosinophils occur in some cases. Neutrophils are uncommon; if prominent, infection should be considered as an alternative or contributing diagnosis. Vasculitis and necrotizing granulomas are absent — features that help distinguish COP from sarcoidosis and granulomatous infections.
Pathogenesis
COP is thought to begin with alveolar epithelial injury — from an infection, drug, toxin, or radiation — that triggers an abnormal repair response. Instead of normal epithelial regeneration, the injured alveolus is filled with a granulation tissue bud that matures incompletely and persists. Bronchoalveolar lavage (BAL) characteristically shows mixed lymphocytosis (CD8+ T cells predominantly, unlike sarcoidosis which is CD4+ dominant), eosinophilia, and foamy macrophages — reflecting alveolar inflammation without a specific pathological agent.
4. Causes and Secondary Triggers
When the OP histological pattern arises from an identifiable cause, it is classified as secondary organizing pneumonia (SOP). A thorough search for secondary causes is essential before diagnosing COP.
Infections
The most common cause of secondary OP. SARS-CoV-2 emerged as a recognized trigger post-2020, with patients developing organizing pneumonia pattern on CT after acute COVID-19 infection, responsive to steroids. Other viral triggers include Influenza, RSV, Adenovirus, CMV, and EBV. Bacterial causes include Mycoplasma pneumoniae (classic), Legionella, and Chlamydophila. Fungal causes include Pneumocystis jirovecii in immunocompromised patients.
Drug-Induced Organizing Pneumonia
Amiodarone is the most commonly implicated drug; amiodarone-induced OP may not fully resolve and can progress even after discontinuation due to the drug's long half-life (~100 days). Other culprit drugs include bleomycin, methotrexate, and immune checkpoint inhibitors (ICI-OP). Pembrolizumab, nivolumab, and atezolizumab cause irAE-OP in 2–5% of patients; the clinical picture of bilateral ground glass and consolidation is important to recognize. Management requires holding the ICI and starting corticosteroids; grade 3–4 toxicity requires permanent ICI discontinuation. Additional implicated agents include cyclophosphamide, nitrofurantoin, and gold therapy.
Connective Tissue Diseases
Rheumatoid arthritis is the most common CTD associated with OP pattern, occurring in approximately 10% of RA-ILD cases. Polymyositis and dermatomyositis — especially clinically amyopathic dermatomyositis (CADM) with anti-MDA5 antibody — are associated with rapidly progressive ILD with an OP/NSIP pattern. Systemic sclerosis, Sjögren's syndrome, and mixed connective tissue disease (MCTD) are additional CTD triggers.
Radiation Pneumonitis
Organizing pneumonia can develop distal to or within the radiation field weeks to months after thoracic radiation therapy. CT shows consolidation in the radiation distribution, or sometimes a migratory or distal pattern. The condition responds well to corticosteroids.
Post-Transplant
Graft-versus-host lung disease in allogeneic stem cell transplant recipients can manifest as organizing pneumonia; prognosis in this setting is generally poor.
Idiopathic (COP)
No identifiable cause is found after thorough workup in approximately 50% of cases, yielding the diagnosis of cryptogenic organizing pneumonia.
5. Clinical Presentation
COP presents subacutely over weeks to a few months, with a typical course of 1–3 months before diagnosis. The clinical picture often mimics community-acquired pneumonia, and the failure to respond to antibiotics is a key diagnostic clue.
Prodrome and Respiratory Symptoms
Most patients describe a flu-like illness (fever, malaise, myalgia) lasting 1–3 weeks, followed by persistent dry cough and progressive dyspnea that do not respond to antibiotics. The cough is typically nonproductive. Progressive dyspnea on exertion and mild to moderate breathlessness are the dominant respiratory complaints. Pleuritic chest pain is less common. Hemoptysis is rare and should raise concern for alternative diagnoses such as malignancy or vasculitis.
Constitutional Symptoms
Low-grade fever is present in 40–50% of patients — a feature that distinguishes COP from IPF/UIP where fever is absent. Fatigue, weight loss of 5–10 kg over weeks to months, and night sweats are common. The constellation of persistent cough, fever, and infiltrates not responding to antibiotics over 4–8 weeks is characteristic and should prompt urgent consideration of COP.
Physical Examination
Bilateral inspiratory crackles (Velcro crackles) are present in 70–75% of patients. Mild tachypnea is common. Importantly, clubbing is absent in COP — its presence is a feature of IPF/UIP, not COP — and this distinction is clinically valuable when evaluating patients with bilateral infiltrates.
Pulmonary Function
COP produces a restrictive pattern on spirometry: reduced FVC, reduced TLC, and a normal or mildly reduced FEV1/FVC ratio. This differs fundamentally from obstructive conditions such as COPD, asthma, and constrictive bronchiolitis. Diffusion capacity (DLCO) is reduced, reflecting alveolar filling and inflammation. Hypoxemia occurs on exertion or at rest in advanced cases.
Laboratory Findings
Elevated ESR and CRP are common but nonspecific. Mild leukocytosis may be present. Serum LDH is mildly elevated in some patients. ANA and rheumatoid factor should be checked to screen for CTD-associated organizing pneumonia.
6. Imaging Findings
High-resolution CT (HRCT) of the chest is critical for diagnosis and shows characteristic patterns that, while not pathognomonic, are highly suggestive of COP in the right clinical context.
Bilateral Peripheral Consolidation
The most common pattern, present in 60–80% of cases. Patchy bilateral airspace consolidation appears in subpleural regions, predominantly in the lower lobes. A peribronchovascular distribution is often present. Ground-glass opacities surround or accompany areas of consolidation. This bilateral, lower-lobe, subpleural consolidation pattern mimics community-acquired pneumonia radiologically.
Reverse Halo Sign (Atoll Sign)
A ring of consolidation surrounding a central area of ground-glass opacity — a "hole" of ground glass inside a ring of denser consolidation. This finding is seen in approximately 19% of COP on CT. Once considered fairly specific for COP, it is also reported in pulmonary infarction, fungal infection, tuberculosis, and lymphoma. When present in the appropriate clinical context, it is a helpful diagnostic clue.
Nodular Form
Multiple small nodules (5–10 mm) distributed along bronchovascular bundles; this pattern can mimic sarcoidosis or metastatic disease and may require tissue sampling to differentiate. FDG-PET/CT is useful in this setting, as OP is PET-avid and can mimic malignancy.
Migratory Consolidation
One of the most characteristic and diagnostically helpful features of COP is the apparent migration of consolidative infiltrates from one region to another on serial CT or chest X-ray, even without treatment. This spontaneous migratory quality is distinctive and should always prompt consideration of COP when observed.
Pleural Effusion
Present in approximately 30% of patients; typically small and bilateral. Unlike malignant effusions, COP-associated pleural effusions do not shift with gravity on decubitus films. Pleural involvement is less marked than in acute eosinophilic pneumonia (AEP).
What COP Does Not Look Like
COP does not produce significant honeycomb change (honeycomb remodeling signals fibrosis and IPF/UIP, not COP) and does not typically cause bilateral hilar lymphadenopathy that predominates (which suggests sarcoidosis). The absence of these features helps narrow the differential diagnosis toward COP.
7. Diagnosis
Diagnosis requires a compatible clinical-radiological picture, exclusion of infection and malignancy, and histological confirmation when transbronchial biopsy is nondiagnostic or when presentation is atypical.
Bronchoscopy with BAL
BAL shows lymphocytosis (typically 20–40% lymphocytes, with a decreased or normal CD4/CD8 ratio reflecting CD8 predominance); mild eosinophilia and neutrophilia may be present; foamy macrophages are present. BAL lymphocytosis supports an inflammatory etiology and is consistent with COP but is not specific. Cultures and special stains are essential to exclude infection before attributing findings to COP.
Transbronchial Lung Biopsy
Shows the OP pattern (Masson bodies) in approximately 70% of COP cases, yielding enough tissue to demonstrate the characteristic buds of granulation tissue. This is often sufficient when the clinical-CT picture is strongly consistent with COP. Risks include pneumothorax (~2%) and bleeding. Diagnostic yield is lower than surgical biopsy, and transbronchial biopsy may not provide sufficient tissue to distinguish COP from other OP-pattern ILDs with confidence in atypical presentations.
Surgical Lung Biopsy (Gold Standard)
Video-assisted thoracoscopic surgery (VATS) biopsy from 2–3 lobes yields definitive diagnosis. Indicated when transbronchial biopsy is nondiagnostic, when clinical presentation is atypical, when CT shows features suggesting an alternative diagnosis (UIP pattern, diffuse alveolar damage), or when CT shows a predominantly nodular or peribronchovascular pattern without classic peripheral consolidation. Histology reveals Masson bodies in alveolar ducts, temporal uniformity, preserved alveolar architecture, and absence of vasculitis or necrotizing granulomas.
Ruling Out Secondary Causes
A systematic search for secondary OP is essential before diagnosing COP. This includes a full medication history (amiodarone, bleomycin, MTX, checkpoint inhibitors — with dates and doses); a comprehensive CTD serology panel (ANA, anti-dsDNA, RF, anti-CCP, SSA/SSB, Scl-70, Jo-1, MDA5, PL-7, PL-12, Mi-2 for myositis antibodies); thorough infection workup (BAL cultures, serology for atypical pathogens, PCP silver stain); radiation history; and stem cell transplant history.
FDG-PET/CT
Useful in nodular COP to distinguish from malignancy. OP is FDG-avid — PET-positive — mimicking lung cancer or lymphoma. A PET-positive consolidative or nodular lesion that resolves with a short steroid trial without biopsy is occasionally acceptable in elderly or frail patients with high surgical risk, though biopsy remains preferred when feasible.
8. Treatment
Corticosteroids are the cornerstone of treatment for COP, and the response is among the most dramatic of any interstitial lung disease.
Corticosteroids
COP responds to corticosteroids dramatically — one of the best responses among all idiopathic interstitial pneumonias. The typical regimen is prednisone 1 mg/kg/day (40–60 mg/day) for 4–8 weeks, followed by clinical and radiological reassessment, then a slow taper over 6–12 months total. A commonly used schedule is: 60 mg for 4 weeks, 40 mg for 4 weeks, 30 mg for 4 weeks, 20 mg for 4 weeks, 10 mg for 4 weeks, 5 mg for 4 weeks, then discontinue. Radiological clearing is typically evident by 4–6 weeks; functional improvement follows closely.
Speed of Response
Most patients notice dramatic symptom improvement within 2–4 weeks of starting prednisone. Fever resolves within days. Radiological improvement begins at 2–4 weeks; complete or near-complete radiological resolution typically occurs in 3–6 months. This speed of response is itself diagnostically confirming — an equally dramatic CT response within weeks essentially excludes IPF/UIP, which does not respond to steroids.
Recurrence
30–50% of patients relapse during or after steroid taper. Importantly, relapse is not a sign of treatment failure or progressive disease — it indicates steroid-dependent disease. Prednisone should be reinitiated at the dose that previously controlled disease, and a slower taper considered on the second course. Most patients achieve long-term remission without permanent disability, even after multiple relapses.
Steroid-Sparing Agents
For recurrent, steroid-dependent, or steroid-intolerant COP, steroid-sparing immunosuppressives are used based on case series data (no RCT evidence in COP specifically). Azathioprine (1–3 mg/kg/day; monitor CBC for myelosuppression; check TPMT enzyme activity before starting) is the most commonly used agent. Mycophenolate mofetil (1.5–3 g/day) and tacrolimus are alternatives. Choice is guided by individual patient comorbidities, tolerability, and prior treatment response.
Checkpoint Inhibitor-Associated Organizing Pneumonia
Withhold the ICI immediately upon diagnosis. Start high-dose prednisone at 1–2 mg/kg/day; most patients respond within 2–4 weeks. Grade 3 (requiring hospitalization and supplemental oxygen) or grade 4 (life-threatening) toxicity mandates permanent ICI discontinuation. For steroid-refractory cases, infliximab or mycophenolate mofetil may be added. Rechallenge with ICI after grade 3–4 irOP carries a high risk of recurrence and is generally not recommended.
Secondary COP Treatment
Treatment of underlying cause plus corticosteroids is the dual approach for secondary organizing pneumonia. For CTD-associated OP, optimize CTD-directed immunosuppression with disease-modifying agents and add corticosteroids for active OP. For RA-ILD-OP specifically, rituximab may benefit both the RA and the ILD. For amiodarone-induced OP, discontinue amiodarone when clinically possible (noting the long half-life of ~100 days, with serum levels persisting for months), and add corticosteroids.
Prophylaxis During Corticosteroid Therapy
All patients on prolonged corticosteroids should receive calcium and vitamin D supplementation. A bisphosphonate (alendronate or risedronate) is indicated for DEXA T-score below −2.5 or for anticipated treatment duration greater than 3 months. PCP prophylaxis with trimethoprim-sulfamethoxazole DS three times weekly is recommended for patients on prednisone greater than 20 mg/day for more than 4 weeks.
9. Prognosis and Recurrence
COP has an excellent prognosis compared with most other idiopathic interstitial pneumonias. Overall 5-year mortality from COP itself is low — estimated below 5% for cryptogenic COP in most published series. Most patients achieve complete or near-complete recovery of lung function. Pulmonary fibrosis is rare in COP, in stark contrast to IPF/UIP where progressive fibrosis is invariable and ultimately fatal.
Key Prognostic Determinants
Cause matters: cryptogenic COP has better prognosis than CTD-associated OP, which depends heavily on the activity and severity of the underlying connective tissue disease; post-infectious OP is often self-limited. Initial severity correlates with course — hypoxemia requiring supplemental oxygen or ICU admission predicts a more severe disease course. Histological subtype is prognostically important: a pure OP pattern carries a good prognosis, whereas an OP pattern overlapping with fibrosing NSIP carries a worse prognosis and warrants close monitoring.
Rapidly Progressive COP
A subset of approximately 5% of patients presents with rapidly progressive COP, manifesting as a near-ARDS syndrome with acute respiratory failure, bilateral extensive infiltrates, and high early mortality. This form requires aggressive ICU management and high-dose intravenous methylprednisolone (typically 500–1000 mg/day for 3–5 days), sometimes with additional immunosuppression.
Distinguishing COP from IPF: The Critical Difference
COP is frequently misdiagnosed as IPF, and this error carries serious consequences because the treatments differ completely. IPF (UIP pattern) causes progressive, irreversible fibrosis; corticosteroids do not help IPF and may cause harm; median survival is 3–5 years from diagnosis. COP causes reversible organizing pneumonia; it responds dramatically to corticosteroids; prognosis is excellent. The CT pattern is the key discriminator: UIP (IPF) shows basal peripheral honeycombing with traction bronchiectasis and a subpleural gradient; COP shows bilateral peripheral consolidation, ground-glass opacities, the reverse halo sign, and no honeycombing. When doubt persists, surgical lung biopsy to distinguish UIP from OP histology is strongly recommended before committing a patient to an IPF management pathway.
10. Recent Research
Post-COVID Organizing Pneumonia
A recognized sequela of SARS-CoV-2 infection emerged prominently after 2020. Patients with persistence of ground-glass and consolidative opacities weeks to months after acute COVID-19, showing an OP pattern on BAL or biopsy, respond to corticosteroids confirming the OP diagnosis. Incidence remains under study and is likely underrecognized; recognition is clinically important as these patients benefit from steroid treatment rather than watchful waiting.
Checkpoint Inhibitor-Associated Organizing Pneumonia (irOP)
As checkpoint inhibitor use in oncology expands dramatically, irOP is increasingly recognized as one of the most common ICI-related lung toxicities. Research is active in identifying predictive biomarkers for irOP risk — including baseline CT features, tumor histology, ICI type and dose — and in optimizing corticosteroid regimens and timing for different severity grades. The clinical overlap between irOP and other ICI pneumonitis patterns (DAD, NSIP, hypersensitivity pneumonitis) is under active investigation.
Nintedanib in COP
Case reports and small series suggest potential benefit from nintedanib (a tyrosine kinase inhibitor approved for IPF and systemic sclerosis-ILD) in COP patients with a fibrosing component or steroid-dependent course. Prospective clinical trial data are lacking; use remains experimental and individual.
Immunosuppressive Combinations for Recurrent COP
Retrospective series examining azathioprine combined with corticosteroids for recurrent COP demonstrate meaningful steroid-dose reduction while maintaining remission, reducing the cumulative steroid burden and associated toxicities. Mycophenolate mofetil shows similar steroid-sparing properties in observational data.
Molecular Biomarkers
BAL lymphocyte subset analysis (CD4/CD8 ratio) and serum KL-6 (Krebs von den Lungen-6) levels are under study as non-invasive biomarkers for monitoring COP disease activity and predicting relapse risk. KL-6 is produced by type II pneumocytes and is elevated in ILD more broadly; its kinetics during COP treatment correlate with radiological response in preliminary studies.
11. Research Papers
Search PubMed for the latest evidence:
- Cryptogenic organizing pneumonia
- Organizing pneumonia BOOP corticosteroid
- Cryptogenic organizing pneumonia CT imaging
- Organizing pneumonia reverse halo sign
- Checkpoint inhibitor organizing pneumonia
- Organizing pneumonia connective tissue disease
- Post-COVID organizing pneumonia
- Cryptogenic organizing pneumonia recurrence
- Organizing pneumonia amiodarone drug-induced
- Organizing pneumonia BAL lymphocytosis
- Organizing pneumonia versus pulmonary fibrosis IPF
- Organizing pneumonia Masson bodies histology
Key Published Studies
- Epler GR, et al. Bronchiolitis obliterans organizing pneumonia. N Engl J Med. 1985;312(3):152–158 — Search PubMed. DOI: 10.1056/NEJM198501173120304
- Cordier JF. Cryptogenic organising pneumonia. Eur Respir J. 2006;28(2):422–446 — Search PubMed. DOI: 10.1183/09031936.06.00013505
- Travis WD, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic interstitial pneumonias: classification, diagnosis, and management. Am J Respir Crit Care Med. 2013;188(6):733–748. PMID: 24032382. DOI: 10.1164/rccm.201308-1483ST
- Lee JW, et al. CT findings in cryptogenic organizing pneumonia. Radiology. 1994;190(1):255–260 — Search PubMed. DOI: 10.1148/radiology.190.1.8259419
- Baque-Juston M, et al. Organizing pneumonia: what is it? A conceptual approach and pictorial review. Diagn Interv Imaging. 2014;95(9):771–777 — Search PubMed. DOI: 10.1016/j.diii.2014.01.004
- Drakopanagiotakis F, et al. Cryptogenic and secondary organizing pneumonia: clinical presentation, radiographic findings, treatment response, and prognosis. Chest. 2011;139(4):893–900 — Search PubMed. DOI: 10.1378/chest.10-1960
- Sveinsson OA, et al. Clinical features in secondary and cryptogenic organising pneumonia. Int J Med Sci. 2007;4(5):272–279 — Search PubMed. DOI: 10.7150/ijms.4.272
- Lazor R, et al. Cryptogenic organizing pneumonia. Characteristics of relapses in a series of 48 patients. Am J Respir Crit Care Med. 2000;162(2):571–577 — Search PubMed. DOI: 10.1164/ajrccm.162.2.9909116
- Sulavik SB. The concept of "organizing pneumonia." Chest. 1989;96(4):967–969 — Search PubMed. DOI: 10.1378/chest.96.4.967
- Khunger M, et al. Incidence of pneumonitis with use of programmed death 1 and programmed death-ligand 1 inhibitors in non-small cell lung cancer. Chest. 2017;152(2):271–281 — Search PubMed. DOI: 10.1016/j.chest.2017.04.177
- Raghu G, et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am J Respir Crit Care Med. 2011;183(6):788–824. PMID: 21471066. DOI: 10.1164/rccm.2009-040GL
- King TE Jr, et al. Idiopathic interstitial pneumonias. Curr Opin Pulm Med. 1998;4(5):272–285 — Search PubMed. DOI: 10.1097/00063198-199809000-00002
12. Connections
- Interstitial Lung Disease
- Pneumonia
- Pulmonary Fibrosis
- Eosinophilic Pneumonia
- Sarcoidosis
- Hypersensitivity Pneumonitis
- Rheumatoid Arthritis
- Lung Cancer
- Lymphangioleiomyomatosis
- All Conditions
- Vitamin D3
- Magnesium