MCAS (Mast Cell Activation Syndrome): History and Discovery
Mast Cell Activation Syndrome (MCAS) is one of the youngest named conditions in this entire encyclopedia, and one of the most genuinely contested. Its building blocks are old — the mast cell itself was discovered and named in 1878, and a disease of too many mast cells (mastocytosis) was mapped out across the following century. But the idea that mast cells could be inappropriately activated — firing off histamine, tryptase, and other mediators throughout the body without the cellular overgrowth of mastocytosis — only crystallized into a named diagnosis around 2007 to 2012. This page traces that long fuse honestly. Where the history is settled it says so; where the diagnosis is still actively debated among the very experts who proposed it, it says that too. Nothing here is a substitute for evaluation by a clinician.
Table of Contents
- Paul Ehrlich and the Discovery of the Mast Cell (1878)
- From Urticaria Pigmentosa to Systemic Mastocytosis
- The KIT Mutation and the Clonal Era
- Activation Without Proliferation: A New Idea
- Naming MCAS: 2007 to 2010
- The 2012 Consensus Criteria
- The Ongoing Debate: Two Schools
- Where Things Stand Today
- Research Papers and References
- Connections
- Featured Videos
Paul Ehrlich and the Discovery of the Mast Cell (1878)
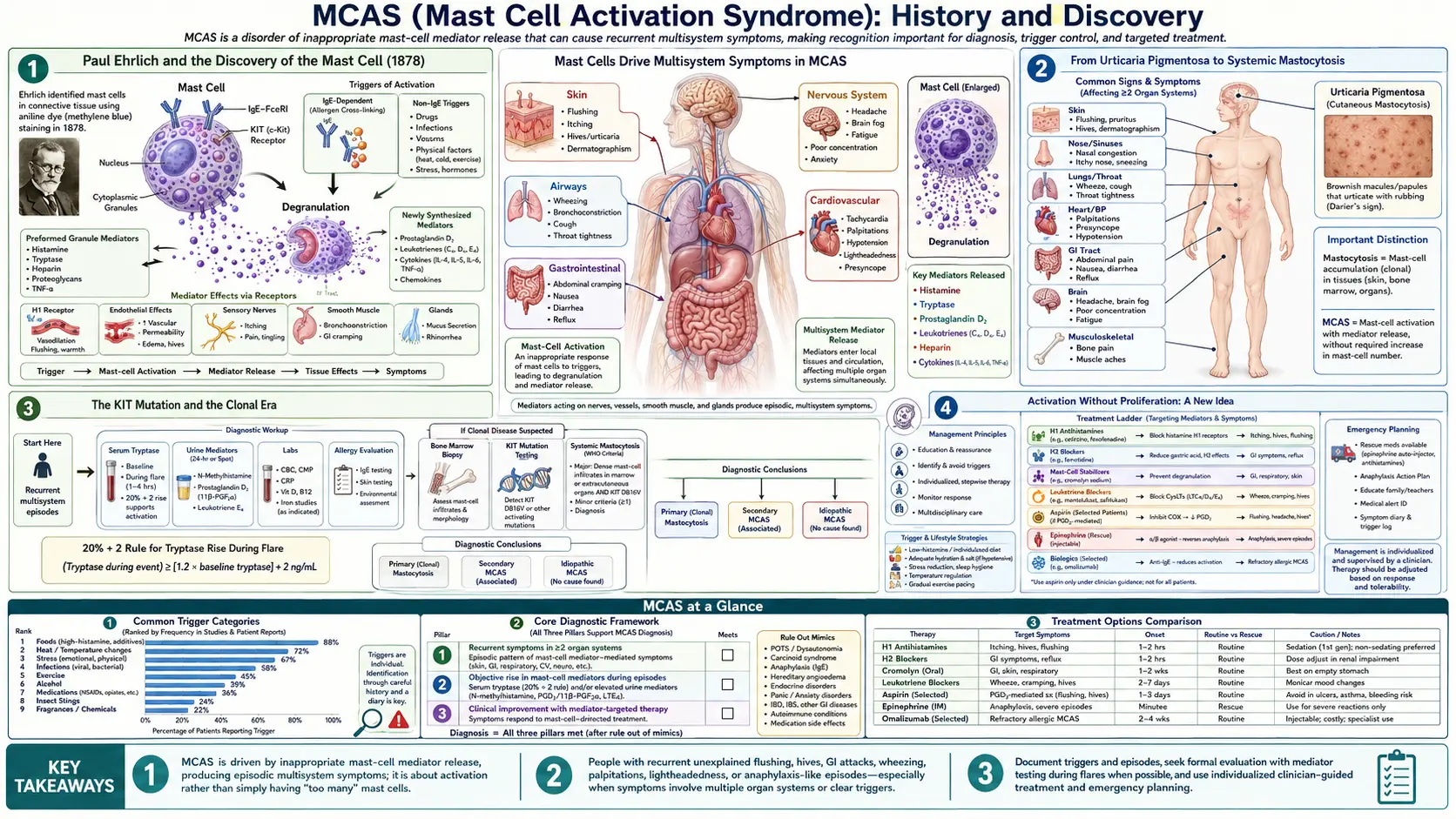
The history of MCAS begins with the cell at its center. On 17 June 1878, a twenty-four-year-old medical student named Paul Ehrlich defended his doctoral thesis at the medical faculty of Leipzig University, titled (in translation) Contributions to the Theory and Practice of Histological Staining. Working with the new aniline dyes of the German chemical industry, Ehrlich noticed a distinctive class of connective-tissue cells whose cytoplasm was crammed with granules that turned a contrasting color — a property later called metachromasia — when stained with basic dyes.
Ehrlich named these cells "Mastzellen", from the German Mast, meaning fattening or well-fed. The granules made the cells look gorged, and he initially — wrongly, as it turned out — supposed they were connective-tissue cells that had taken up surrounding nutrients. We now know the granules are pre-formed packages of inflammatory mediators, chiefly histamine and the enzyme tryptase, along with heparin and many others. The English term "mast cell" is a direct translation of Ehrlich's coinage, and it has stuck for nearly a century and a half.
For decades the mast cell was a histological curiosity in search of a job. Its central role in allergy and anaphylaxis — releasing its granule contents when the antibody IgE on its surface meets an allergen — was worked out only across the twentieth century, long after Ehrlich's death in 1915. That sequence matters for this story: the cell was named in 1878, its allergic function was understood by the mid-1900s, but the idea that it could misbehave as a distinct named syndrome had to wait until the twenty-first century.
From Urticaria Pigmentosa to Systemic Mastocytosis
The first mast-cell disease recognized was one of too many mast cells in the skin. In 1869, the English physicians Edward Nettleship and Warren Tay described a patient with a chronic, spotty, hive-like skin eruption — the lesions we now associate with cutaneous mastocytosis. The descriptive name "urticaria pigmentosa" (pigmented hives) was coined a few years later, in 1878, by the dermatologist Alfred Sangster. Shortly after Ehrlich described the mast cell, pathologists found that these skin lesions were packed with focal accumulations of exactly that cell type, linking the visible rash to the newly named cell.
For the next several decades mastocytosis was thought of as a skin condition. That changed in 1949, when the American pathologist J. M. Ellis reported an autopsy of a one-year-old infant who had had urticaria pigmentosa from birth along with gastrointestinal symptoms. Ellis found mast-cell infiltrates not only in the skin but throughout the internal organs — liver, spleen, bone marrow, lymph nodes, and more. This was the first clear description of systemic mastocytosis: the recognition that the mast-cell overgrowth could be a body-wide disease, not merely a skin rash.
The defining feature of mastocytosis, then and now, is proliferation — an abnormal increase in the number of mast cells, which accumulate in tissues and can be counted, biopsied, and seen under the microscope. Keep that word in mind. The central conceptual move that later produced MCAS was to ask a different question entirely: what if the problem is not how many mast cells a person has, but how badly the normal number of them is behaving?
The KIT Mutation and the Clonal Era
The molecular turning point for mast-cell disease came in the 1990s. Mast cells depend for their growth and survival on a cell-surface receptor called KIT (also written c-KIT or CD117), which responds to a signal called stem cell factor. In 1993, Furitsu and colleagues identified an activating mutation in the KIT gene — a single amino-acid change designated D816V — in a human mast-cell leukemia cell line (HMC-1). In 1995, Nagata and colleagues found the same KIT D816V mutation in tissue from patients with systemic mastocytosis.
This was a major advance. D816V is a "gain-of-function" mutation: it switches the KIT receptor permanently on, telling the mast cell to grow and survive even when it shouldn't. The mutation is now recognized as the molecular hallmark of most adult systemic mastocytosis, is one of the formal World Health Organization diagnostic criteria, and is the basis for the term clonal — meaning the mast cells share an identifiable, abnormal genetic signature marking them as descendants of a single rogue founder cell. (Clonality can also be flagged by aberrant surface markers such as CD25 on the mast cells.)
The clonal era gave doctors something solid to test for: a mutation, a marker, a measurable abnormality. It sharpened mastocytosis into a precisely definable disease. But it also threw a puzzle into sharp relief. Clinicians kept seeing patients who had all the symptoms of mast-cell mediator release — flushing, hives, abdominal cramping, low blood pressure, even anaphylaxis — yet who did not have enough mast cells to meet mastocytosis criteria, and often did not carry the clonal markers. If it wasn't mastocytosis, what was it?
Activation Without Proliferation: A New Idea
The conceptual heart of MCAS is the distinction between proliferation and activation. Mastocytosis is a disease of proliferation: too many mast cells. The proposed entity of MCAS is a disease of activation: a normal (or near-normal) number of mast cells that are inappropriately and repeatedly triggered to degranulate — to release their stored mediators — producing recurrent, multi-system symptoms. A useful analogy: mastocytosis is having too many smoke alarms installed in the house; MCAS is the idea that a normal number of smoke alarms keep going off when there is no fire.
When a mast cell activates, it releases a chemical cocktail. The best known is histamine, which drives itching, hives, flushing, runny nose, and stomach acid. The enzyme tryptase is released too and is clinically important precisely because a transient rise in blood tryptase can be measured in a laboratory — making it the closest thing the field has to an objective fingerprint of an activation event. Other mediators include prostaglandins, leukotrienes, heparin, and a long list of cytokines, which between them can plausibly touch the skin, gut, lungs, heart and blood vessels, and nervous system — explaining why a true mast-cell activation disorder would, by its nature, be multi-system.
The intellectual groundwork for naming such a disorder was the recognition, in the 1990s and 2000s, that mast-cell activation exists on a spectrum independent of mast-cell numbers. A person could, in principle, have clonal mast cells that activate too easily (a mast-cell disorder lying just short of full mastocytosis), or non-clonal mast cells that activate for reasons not yet understood. Carving a named diagnosis out of that spectrum — deciding where ordinary allergy ends and a distinct "activation syndrome" begins — is exactly the step that proved, and remains, difficult and debated.
Naming MCAS: 2007 to 2010
The term mast cell activation syndrome entered the medical literature in the late 2000s, roughly around 2007, built on accumulating reports from several mast-cell research groups. A pivotal observation was that a subset of patients carrying a diagnosis of "idiopathic anaphylaxis" — severe allergic-type reactions with no identifiable trigger — were found, on careful testing, to harbor small clonal mast-cell populations with abnormal markers. Work in this area by Cem Akin, Dean D. Metcalfe and colleagues at the U.S. National Institutes of Health, together with European groups, suggested these patients occupied a space between ordinary allergy and overt mastocytosis — a clonal mast-cell activation state without enough cells to qualify as mastocytosis. This became known as monoclonal mast cell activation syndrome (MMAS).
The defining formal step came in December 2010, when Cem Akin, Peter Valent, and Dean D. Metcalfe published Mast cell activation syndrome: proposed diagnostic criteria in the Journal of Allergy and Clinical Immunology. This paper crystallized MCAS as a named diagnostic concept and laid out the logic that would define it: (1) typical recurrent symptoms of mast-cell mediator release affecting two or more organ systems; (2) objective laboratory evidence of mediator release, anchored on a transient rise in serum tryptase; and (3) a clinical response to medications that block mast-cell mediators (such as antihistamines). Critically, the criteria required that mastocytosis and other defined disorders be ruled out first.
It is worth being precise about authorship and priority, because this is the kind of claim a history page must get right. The 2010 JACI paper by Akin, Valent, and Metcalfe is the most widely cited proposal of formal diagnostic criteria for MCAS; the underlying idea and the term itself had been taking shape in the literature over the preceding few years. These three researchers, with their collaborators, are the figures most consistently associated with defining the entity.
The 2012 Consensus Criteria
Recognizing that a new diagnosis used inconsistently would cause confusion, an international group of specialists in allergy, immunology, dermatology, and hematology convened a working conference in September 2010 to standardize definitions. The result was published as Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal, led by Peter Valent with Cem Akin, Dean D. Metcalfe, and a panel of international co-authors, appearing in the International Archives of Allergy and Immunology in 2012.
This consensus did two lasting things. First, it proposed a now widely-cited laboratory threshold for confirming an activation event: during a symptomatic episode, the serum total tryptase should rise by at least 20% above the person's own baseline, plus an additional 2 ng/mL. (Worked example from the paper: if baseline tryptase is 10 ng/mL, the level during an event should exceed roughly 14 ng/mL.) Tying the diagnosis to the patient's own baseline, rather than a fixed cutoff, was a deliberate attempt to make the criterion objective and reproducible — and it remains a reference standard for severe systemic activation.
Second, the consensus laid out a classification of MCAS into three forms, a framework still in use:
- Primary (clonal) MCAS — the patient has clonal mast cells (KIT D816V and/or aberrant CD25) but does not meet full mastocytosis criteria; this includes monoclonal MCAS.
- Secondary MCAS — activation driven by an identifiable underlying condition, classically an IgE-mediated allergy or another inflammatory disorder.
- Idiopathic MCAS — the criteria for activation are met, but no clonal marker and no triggering disease can be found.
The Ongoing Debate: Two Schools
Here the history must be told carefully, because MCAS is a genuinely contested diagnosis and it would be misleading to present it as settled. The disagreement is not really about whether mast cells can be inappropriately activated — they can — but about how broadly the label should be applied and how strict the evidence must be. Two broad schools have emerged.
The first, often called the consensus or ECNM/AIM approach (associated with Valent, Akin, Metcalfe and their networks), keeps the requirement for objective biochemical evidence — above all the tryptase rise of 20% + 2 ng/mL — at the center of diagnosis. On this view, the well-defined, defensible territory of MCAS is the monoclonal/primary form and clearly mediator-proven cases. These authors worry that loosening the laboratory requirement risks labeling large numbers of people with non-specific multi-system symptoms as having a mast-cell disease they cannot demonstrate. This stricter framework was reinforced in a later statement sometimes referred to as "consensus-1."
The second, broader school — associated especially with Lawrence Afrin, Gerhard Molderings and colleagues, and articulated in a 2020 proposal sometimes called "consensus-2" — argues that the strict tryptase criterion misses many real patients, because tryptase is often not elevated in milder or more localized activation, and a blood sample frequently cannot be drawn within the narrow window after an episode. This group advocates a wider clinical definition and a longer menu of measurable mediators (such as urinary prostaglandins and histamine metabolites). Critics counter that the broader alternative criteria have low diagnostic specificity — that is, they tend to capture symptoms common in the general population and risk over-diagnosis, potentially crowding out other treatable explanations. Both "over-diagnosed" and "under-diagnosed" positions are argued in the peer-reviewed literature, and that tension is unresolved as of this writing.
Where Things Stand Today
What can be stated with confidence is a relatively short list. The mast cell is real and well characterized. Clonal mast-cell disease short of mastocytosis — monoclonal/primary MCAS — is a recognized, testable entity. The tryptase rise of 20% + 2 ng/mL is an accepted objective marker of a significant systemic activation event. Severe, recurrent, multi-system mast-cell mediator symptoms are a real clinical problem for the patients who have them. None of that is in dispute.
What remains genuinely uncertain is the boundary and the breadth: how often non-clonal "idiopathic" MCAS truly accounts for diffuse, hard-to-pin-down symptoms; which mediators and thresholds should count; and how to separate a real mast-cell disorder from the many other conditions that produce overlapping symptoms. Researchers have noted that MCAS frequently travels alongside conditions such as postural orthostatic tachycardia syndrome (POTS), hypermobile Ehlers-Danlos syndrome, and chronic-fatigue presentations — an intriguing clustering whose mechanism is, at present, a hypothesis under investigation rather than an established causal link. It should be read as a question the field is actively studying, not a settled fact.
The honest summary is this: in less than two decades MCAS has gone from an unnamed clinical puzzle to a defined diagnosis with consensus criteria — and simultaneously to one of the more debated labels in modern medicine. For a patient, the practical takeaway is to seek evaluation from a clinician experienced in mast-cell disease, to pursue objective testing where possible, and to be wary both of having genuine symptoms dismissed and of accepting an MCAS label that forecloses the search for other explanations. The story of MCAS is still being written, and this page will be most useful if it represents that uncertainty faithfully.
Research Papers and References
The references below combine the landmark peer-reviewed papers that defined the mast cell and mast-cell disease with curated PubMed topic-search links into the historical and diagnostic literature. Foundational historical sources — Paul Ehrlich's 1878 doctoral thesis, the 1869 description by Nettleship and Tay, and J. M. Ellis's 1949 report — are named in the article as historical primary sources. Where a stable identifier is available a DOI or PMID link is given; otherwise a PubMed topic search is provided. Each link opens in a new tab.
- Akin C, Valent P, Metcalfe DD. Mast cell activation syndrome: proposed diagnostic criteria. Journal of Allergy and Clinical Immunology. 2010;126(6):1099-1104. — doi:10.1016/j.jaci.2010.08.035
- Valent P, Akin C, Arock M, et al. Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. International Archives of Allergy and Immunology. 2012;157(3):215-225. — doi:10.1159/000328760
- Valent P, Akin C, Bonadonna P, et al. Proposed diagnostic algorithm for patients with suspected mast cell activation syndrome. Journal of Allergy and Clinical Immunology: In Practice. 2019;7(4):1125-1133. — doi:10.1016/j.jaip.2019.01.006
- Afrin LB, Ackerley MB, Bluestein LS, et al. Diagnosis of mast cell activation syndrome: a global "consensus-2." Diagnosis (Berlin). 2021;8(2):137-152. — doi:10.1515/dx-2020-0005
- Valent P, Hartmann K, Bonadonna P, et al. Mast cell activation syndromes: collegium internationale allergologicum update 2022. International Archives of Allergy and Immunology. — PubMed: Valent MCAS criteria update
- Crawford ES, et al. Mast cell activation syndrome: importance of consensus criteria and call for research. Journal of Allergy and Clinical Immunology. 2019. — PMID: 29928922
- Crisis of diagnosis — MCAS overdiagnosed or underdiagnosed? Journal of Allergy and Clinical Immunology: In Practice. 2024. — PubMed: MCAS overdiagnosed or underdiagnosed
- Low diagnostic specificity of proposed alternatives to consensus MCAS criteria. Journal of Allergy and Clinical Immunology. 2025. — PubMed: MCAS criteria diagnostic specificity
- Nagata H, Worobec AS, Oh CK, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients with mastocytosis. Proceedings of the National Academy of Sciences. 1995;92(23):10560-10564. — doi:10.1073/pnas.92.23.10560
- Furitsu T, Tsujimura T, Tono T, et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line (HMC-1). Journal of Clinical Investigation. 1993;92(4):1736-1744. — doi:10.1172/JCI116761
- Crivellato E, Beltrami CA, Mallardi F, Ribatti D. Paul Ehrlich's doctoral thesis: a milestone in the study of mast cells. British Journal of Haematology. 2003;123(1):19-21. — doi:10.1046/j.1365-2141.2003.04573.x
- Ellis JM. Urticaria pigmentosa: a report of a case with autopsy. Archives of Pathology. 1949. (historical systemic mastocytosis report) — PubMed: Ellis systemic mastocytosis history
- History of mast-cell research from Ehrlich's Mastzellen to modern immunology — PubMed: history of mast cell research
- Monoclonal mast cell activation syndrome and clonal markers in idiopathic anaphylaxis — PubMed: monoclonal MCAS and idiopathic anaphylaxis
External Authoritative Resources
- The Mast Cell Disease Society — Mast Cell Activation Syndromes overview
- NIH / NIAID — Mastocytosis and mast-cell disorders
- PubMed — All research on mast cell activation syndrome
Connections
- Neurology
- Mast Cell Activation Syndrome (MCAS) — Main Page
- All Conditions
- POTS (Postural Orthostatic Tachycardia Syndrome)
- ME / Chronic Fatigue Syndrome
- Allergies
- Mastocytosis — the older proliferative mast-cell disease whose century of study set the stage for MCAS.
- Alpha-Gal Syndrome