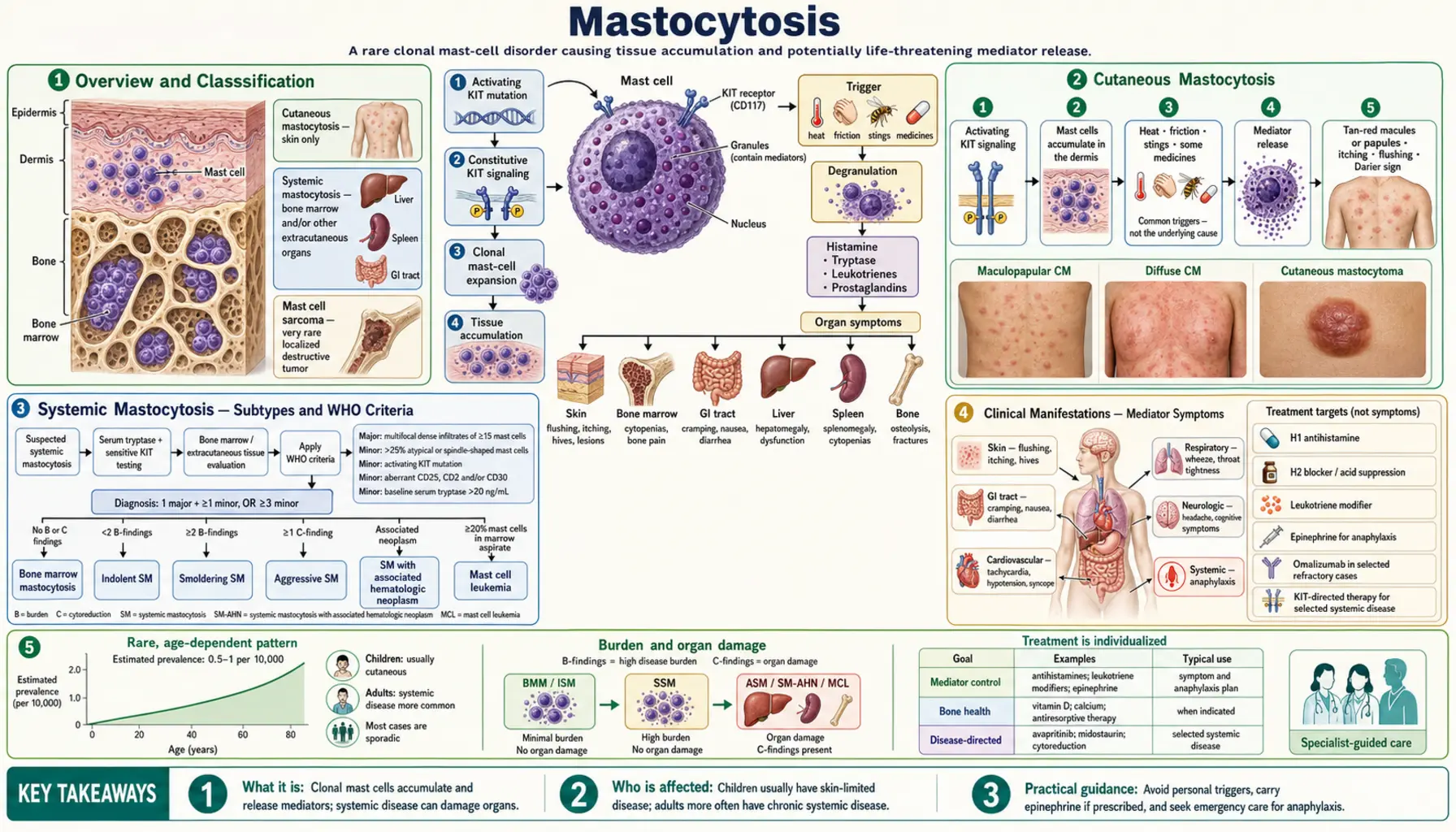
Mastocytosis
Mastocytosis is a clonal mast cell disorder caused by an activating mutation in the KIT gene (most commonly KIT D816V), which encodes the stem cell factor receptor CD117. This mutation drives ligand-independent signaling, resulting in uncontrolled mast cell proliferation and accumulation in one or more organ systems. The condition spans a wide spectrum from skin-limited disease in children to aggressive systemic forms in adults requiring targeted kinase inhibitor therapy.
Interactive Visualization Histamine, Mast Cells & Allergy — trigger a mast cell yourself Arm a mast cell with IgE, let the allergen cross-link it, and watch histamine burst out to raise a wheal — then block the H1 receptor and see symptoms fall while histamine still pours out. Launch →Table of Contents
- Overview and Classification
- Cutaneous Mastocytosis
- Systemic Mastocytosis — Subtypes and WHO Criteria
- Clinical Manifestations — Mediator Symptoms
- Bone Manifestations and Osteoporosis
- Diagnosis — Laboratory and Pathology
- Treatment — Symptomatic Management
- Treatment — Advanced Systemic Mastocytosis
- Hereditary Alpha-Tryptasemia
- Key Research Papers
- Connections
- Featured Videos
Overview and Classification
Mastocytosis arises from a clonal expansion of mast cells carrying an activating mutation, most commonly KIT D816V — a single amino acid substitution (aspartate to valine at position 816) in the activation loop of the KIT tyrosine kinase receptor. This substitution renders the receptor constitutively active without requiring stem cell factor (SCF) ligand binding, driving autonomous mast cell proliferation.
The KIT gene encodes CD117, the stem cell factor receptor expressed on mast cells, hematopoietic progenitors, and melanocytes. In normal physiology, SCF binding induces receptor dimerization and downstream signaling through RAS/MAPK and PI3K/AKT pathways. The D816V mutation bypasses this requirement. This mutation is found in over 90% of adult systemic mastocytosis cases and in a significant proportion of pediatric cases.
The two major categories of mastocytosis are:
- Cutaneous Mastocytosis (CM): mast cell accumulation confined to the skin; predominantly affects children; most cases resolve by puberty
- Systemic Mastocytosis (SM): mast cell infiltration of extracutaneous organs, most critically the bone marrow; predominantly affects adults; requires specific WHO diagnostic criteria for classification and subtyping
Cutaneous Mastocytosis
Cutaneous mastocytosis is the most common form of mastocytosis in children and is characterized by mast cell infiltration of the dermis without systemic organ involvement.
Urticaria Pigmentosa (Maculopapular Cutaneous Mastocytosis)
Urticaria pigmentosa is the most frequent form of CM, also termed maculopapular cutaneous mastocytosis (MPCM). It presents as tan or reddish-brown macules and papules distributed across the trunk, extremities, and occasionally the face. These lesions represent focal dermal accumulations of mast cells. In children, lesions tend to be fewer and larger; in adults (who can develop CM as a variant of systemic disease), lesions are typically more numerous and smaller.
Darier's Sign
Darier's sign is the pathognomonic physical finding in mastocytosis. When a skin lesion is firmly stroked, the mechanical stimulation triggers mast cell degranulation, releasing histamine and other mediators locally. This produces a localized wheal-and-flare reaction (urticaria) overlying the stroked lesion within minutes. A positive Darier's sign strongly supports the diagnosis of cutaneous mastocytosis and distinguishes mast cell infiltrates from other pigmented skin lesions such as lentigines or melanocytic nevi.
Pediatric Course and Prognosis
Childhood cutaneous mastocytosis carries an excellent prognosis. The majority of children with CM — particularly those with onset in infancy — experience spontaneous regression of skin lesions by puberty as mast cell burden naturally decreases. Systemic involvement is minimal in pure CM. Parents should be educated about mast cell triggers (heat, friction, certain medications) that can provoke mediator release and symptomatic episodes, including flushing and, rarely, anaphylaxis.
Other CM Subtypes
- Diffuse cutaneous mastocytosis (DCM): rare, occurs in infants; widespread mast cell infiltration of the entire skin surface; can cause extensive blistering (bullous formation) due to massive mast cell degranulation; more clinically severe than MPCM
- Solitary mastocytoma: a single mast cell tumor nodule, usually appearing in the first months of life; most resolve spontaneously; generally excellent prognosis; biopsy confirms the diagnosis if needed
Systemic Mastocytosis — Subtypes and WHO Criteria
Systemic mastocytosis is defined by clonal mast cell infiltration of extracutaneous organs. The bone marrow is the primary site of involvement and the key diagnostic organ. SM is predominantly a disease of adults; the KIT D816V mutation is present in the vast majority of cases.
WHO 2022 Diagnostic Criteria
The diagnosis of SM requires fulfillment of either 1 major criterion plus 1 minor criterion, or at least 3 minor criteria:
Major criterion:
- Multifocal dense mast cell aggregates (clusters of 15 or more mast cells) in bone marrow biopsy sections and/or in sections of other extracutaneous organs
Minor criteria:
- More than 25% of the mast cells in the biopsy are spindle-shaped or have atypical morphology
- Detection of an activating point mutation at codon 816 of KIT (typically D816V) in bone marrow, blood, or other extracutaneous organ
- Mast cells in bone marrow, blood, or other extracutaneous organ express CD25 with or without CD2, in addition to normal mast cell markers (CD117, tryptase)
- Serum total tryptase persistently exceeds 20 ng/mL (does not apply if there is an associated clonal myeloid neoplasm)
SM Subtypes (in Order of Increasing Severity)
- Indolent SM (ISM): fulfills SM criteria; no C-findings (organ damage); no evidence of associated hematologic neoplasm; normal peripheral blood counts; best prognosis among SM subtypes; near-normal life expectancy; symptoms driven primarily by mediator release rather than organ infiltration
- Smoldering SM (SSM): higher mast cell burden than ISM (often serum tryptase >200 ng/mL, or >30% mast cells on BM biopsy, or splenomegaly without C-findings); no frank organ damage yet; intermediate risk of progression
- SM with Associated Hematologic Neoplasm (SM-AHN): formerly called SM-AHNMD; SM occurs alongside a separate clonal myeloid or lymphoid neoplasm (most commonly chronic myelomonocytic leukemia, myelodysplastic syndrome, or acute myeloid leukemia); prognosis driven largely by the associated neoplasm
- Aggressive SM (ASM): presence of one or more C-findings indicating organ damage attributable to mast cell infiltration (see below); requires cytoreductive therapy
- Mast Cell Leukemia (MCL): the rarest and most severe form; defined by circulating mast cells comprising 20% or more of the white cell differential on peripheral blood smear; alternatively, >20% mast cells on bone marrow aspirate smear regardless of peripheral blood; median survival historically measured in months; currently treated with avapritinib
C-Findings (Organ Damage Markers)
C-findings represent end-organ damage attributable to mast cell infiltration and define the threshold between smoldering/indolent and aggressive disease:
- Absolute neutrophil count below 1.0 × 10&sup9;/L
- Hemoglobin below 10 g/dL
- Platelet count below 100 × 10&sup9;/L
- ALT or AST elevated more than 3-fold above the upper limit of normal with or without ascites
- Serum albumin below 3.5 g/dL
- Pathologic fractures due to mast cell-induced osteolysis
- Large liver or spleen with portal hypertension and impaired liver function
- Malabsorption with weight loss due to GI mast cell infiltration
Clinical Manifestations — Mediator Symptoms
A defining feature of mastocytosis — across all subtypes — is the release of preformed and newly synthesized mast cell mediators. These episodes can occur spontaneously or be provoked by identifiable triggers. The main mediators involved include histamine, tryptase, prostaglandin D2, heparin, leukotrienes (LTC4, LTD4), and cytokines (IL-6, TNF-alpha).
Flushing
Episodic flushing — sudden reddening and warmth of the face, neck, and upper chest — is one of the most common presenting symptoms. Flushing in mastocytosis is driven primarily by histamine and prostaglandin D2 rather than the catecholamine-mediated flushing of pheochromocytoma. Common triggers include:
- Heat (hot baths, hot weather, exercise)
- Alcohol — even small quantities
- Emotional stress
- NSAIDs (especially aspirin, which can trigger massive prostaglandin-related flushing)
- Opioid analgesics (particularly codeine and morphine, which directly stimulate mast cell degranulation)
- Iodinated contrast media (intravenous dye)
- Hymenoptera venom (bee and wasp stings)
- Physical friction to skin lesions
Urticaria and Pruritus
Generalized or localized itching is nearly universal in cutaneous disease and common in SM. Mechanical stimulation (Darier's sign) can provoke hive formation at lesion sites. Aquagenic urticaria (itching triggered by water contact) also occurs in some patients.
Gastrointestinal Symptoms
GI mast cell accumulation produces multiple effects. Histamine stimulates gastric acid hypersecretion via H2 receptors on parietal cells, leading to peptic ulcer disease, epigastric pain, and reflux. Mast cell infiltration of the intestinal wall causes cramping, bloating, diarrhea, and in severe cases malabsorption with weight loss. Abdominal pain is among the most debilitating symptoms for many patients with SM and frequently requires multi-drug management.
Neurological and Cognitive Symptoms
Headache, difficulty concentrating, and the constellation of symptoms collectively termed "brain fog" are reported frequently. These symptoms may be histamine-mediated (histamine crosses the blood-brain barrier and modulates neurological function via H1 and H3 receptors) or related to prostaglandin and cytokine release. Depression and anxiety are also increased in mastocytosis populations.
Anaphylaxis
Anaphylaxis — systemic, potentially life-threatening mast cell degranulation — is a major concern in all SM patients. Several features are distinctive:
- SM patients, including those with indolent disease, have a paradoxically high rate of severe anaphylaxis relative to their overall disease burden
- Hymenoptera venom (bee, wasp, yellow jacket) is the classic and most dangerous trigger; venom-triggered anaphylaxis in mastocytosis can be fatal without rapid epinephrine administration
- Anaphylaxis may also occur without an identifiable trigger (idiopathic)
- Standard anaphylaxis symptoms apply: urticaria, angioedema, bronchospasm, hypotension, loss of consciousness
- All patients with SM must carry and be trained to use two epinephrine auto-injectors (EpiPens) at all times
Importantly, serum tryptase measured 1-3 hours after an anaphylactic episode is markedly elevated in virtually all SM patients (due to the large mast cell burden); a persistently elevated tryptase weeks after the acute event suggests SM as the underlying diagnosis.
Bone Manifestations and Osteoporosis
Skeletal disease is a major and underrecognized complication of systemic mastocytosis, even in the indolent subtype. Mast cells release multiple mediators that directly perturb bone remodeling:
- Histamine stimulates osteoclast activity via H1 and H2 receptors
- Heparin promotes osteoclast differentiation and inhibits osteoblast function
- Tryptase activates protease-activated receptors on bone cells
- IL-6 and TNF-alpha increase RANKL expression, further driving osteoclast-mediated bone resorption
Osteoporosis
Osteoporosis occurs at significantly higher rates in SM patients compared to age-matched controls. Fractures — including vertebral compression fractures — are common and can be the presenting manifestation of previously undiagnosed SM. The lumbar spine is most frequently affected. Osteoporosis in SM can be severe even in young patients and in the absence of other C-findings, making bone density monitoring essential for all SM patients regardless of subtype.
A DEXA (dual-energy X-ray absorptiometry) scan is recommended at the time of diagnosis for all SM patients. Bisphosphonate therapy (alendronate orally, or zoledronate intravenously for severe cases) is indicated for patients with low bone mineral density (T-score below -2.5) or with prevalent fragility fractures. Annual DEXA reassessment is appropriate during treatment. Calcium and vitamin D supplementation are standard adjuncts.
Osteosclerosis
A paradoxical increase in bone density — osteosclerosis — occurs in a subset of SM patients, particularly affecting the axial skeleton (spine, pelvis). This results in abnormally dense, thickened trabeculae on imaging. Patients may simultaneously have osteosclerotic regions alongside osteoporotic regions, creating a mixed lytic/sclerotic pattern on radiographs or CT scans. Osteosclerosis is more common in patients with higher mast cell burden and advanced SM subtypes. Despite the radiographic density, structural bone quality is abnormal and fracture risk may still be elevated.
Radiologic Assessment
Plain radiographs of the spine and pelvis can identify mixed lytic/sclerotic changes. CT provides higher resolution detail of bone architecture. A nuclear medicine bone scan (technetium-99m) can help identify sites of active bone turnover. MRI of the spine is sensitive for detecting mast cell infiltration of vertebral marrow (hypointense on T1, variable on T2) and associated marrow edema.
Diagnosis — Laboratory and Pathology
Diagnosis of mastocytosis integrates clinical assessment, laboratory studies, imaging, and tissue biopsy. A systematic approach is essential given the heterogeneity of presentations.
Serum Tryptase
Serum total tryptase is the most clinically useful biomarker for mastocytosis. Tryptase is a serine protease stored in mast cell secretory granules. In healthy individuals, baseline serum tryptase is typically below 11.4 ng/mL. Key interpretive points:
- A persistently elevated baseline tryptase above 20 ng/mL (measured at least 24 hours after any acute event) is a minor diagnostic criterion for SM
- Tryptase levels are proportional to total body mast cell burden and correlate with SM subtype severity
- Transient elevation during anaphylaxis (peak at 1-3 hours, returning to baseline within 24 hours) contrasts with the persistent elevation seen in SM
- Hereditary alpha-tryptasemia (HαT) can cause elevated baseline tryptase without mastocytosis and must be distinguished through genetic testing
KIT D816V Mutation Testing
Detection of KIT D816V is a minor diagnostic criterion and critical for treatment selection. Testing options include:
- Peripheral blood allele-specific PCR: highly sensitive modern assays can detect the mutation in blood in the majority of SM patients, enabling a less invasive initial screen
- Bone marrow PCR: more sensitive than peripheral blood; particularly important when blood-based testing is negative but clinical suspicion remains high
- KIT D816V positivity also determines eligibility for avapritinib therapy in advanced SM
Bone Marrow Biopsy
Bone marrow trephine biopsy is required to fulfill the major diagnostic criterion for SM and to assess disease burden. A comprehensive biopsy evaluation includes:
- Hematoxylin and eosin (H&E) staining for overall morphology and detection of mast cell aggregates
- Giemsa staining for mast cell granule visualization
- Immunohistochemistry: CD117 (marks mast cells), tryptase (confirms mast cell identity), CD25 (aberrant expression in clonal mast cells — not present on normal mast cells), CD2 (variable aberrant expression)
- The finding of multifocal aggregates of 15 or more mast cells is the major diagnostic criterion
Skin Biopsy
If urticaria pigmentosa lesions are present, punch biopsy of a representative lesion confirms dermal mast cell infiltration. Staining with tryptase or CD117 highlights the accumulated mast cells. A positive skin biopsy in the appropriate clinical context (with Darier's sign) strongly supports the diagnosis and may spare some patients the need for immediate bone marrow evaluation.
Additional Laboratory Studies
- Complete blood count with differential: cytopenias (low neutrophils, anemia, thrombocytopenia) indicate C-findings; eosinophilia may accompany SM-AHN
- Comprehensive metabolic panel: liver function tests for hepatic involvement; albumin level
- 24-hour urine N-methylhistamine: elevated in active mast cell disease; useful for monitoring mediator burden
- Urinary prostaglandin D2 metabolites (11-beta-PGF2-alpha): elevation indicates active prostaglandin D2 release
- CT scan of chest/abdomen/pelvis: assesses for organomegaly (liver, spleen, lymph nodes), lymphadenopathy, ascites
- DEXA scan: bone mineral density baseline at diagnosis
Treatment — Symptomatic Management
Symptom control targeting mast cell mediator effects is the foundation of treatment for all mastocytosis patients, including those with CM and indolent SM who do not require cytoreductive therapy.
Antihistamines
Antihistamine therapy is first-line and should be initiated in all symptomatic patients:
- H1 antihistamines (cetirizine, loratadine, fexofenadine): control flushing, urticaria, pruritus, and reduce anaphylaxis risk; non-sedating agents preferred for daily use; higher doses than standard allergy dosing are often required
- H2 antihistamines (famotidine, ranitidine): reduce gastric acid hypersecretion, control GI symptoms, and provide additional systemic H1-type symptom coverage; ideally taken 30 minutes before meals
Epinephrine Auto-Injector
All patients with systemic mastocytosis — and any CM patient with a history of anaphylaxis or significant mediator symptoms — must be prescribed and carry two epinephrine auto-injectors (EpiPen 0.3 mg or equivalent). Two devices are prescribed because a single dose may be insufficient for severe anaphylaxis in SM patients given their high mast cell burden. Patients, family members, and caregivers must receive training on recognition of anaphylaxis and injection technique. Liberal use should be encouraged: it is safer to use epinephrine unnecessarily than to delay its use during anaphylaxis.
Cromolyn Sodium
Oral cromolyn sodium (disodium cromoglycate) is a mast cell stabilizer taken four times daily before meals and at bedtime. It is poorly absorbed from the gastrointestinal tract, meaning its primary action is local stabilization of intestinal mast cells. It is effective for abdominal cramping, diarrhea, bloating, and abdominal pain in SM. It has minimal systemic side effects given its poor absorption.
Bisphosphonates and Bone Protection
- Oral bisphosphonates (alendronate 70 mg weekly): first-line for SM patients with osteoporosis (T-score < -2.5) or osteopenia with fragility fracture
- Intravenous zoledronate (5 mg annually): preferred for patients with severe osteoporosis, poor oral medication tolerance, or renal considerations within safe limits
- Calcium (1,000-1,200 mg daily) and vitamin D3 (1,000-2,000 IU daily) supplementation are standard adjuncts
- Annual DEXA scan monitoring is appropriate during treatment
Trigger Avoidance
Patient education about personal triggers is essential. Key avoidances include:
- NSAIDs: aspirin and other NSAIDs can trigger massive prostaglandin D2-mediated flushing and anaphylaxis in susceptible SM patients; avoid unless the patient has previously demonstrated good tolerance
- Opioids: codeine and morphine directly stimulate mast cell degranulation through a non-IgE mechanism; fentanyl has substantially less mast cell degranulating activity and is relatively safer for procedural analgesia in mastocytosis patients
- Iodinated contrast media: premedicate with H1 antihistamine, H2 antihistamine, and corticosteroid before CT or angiographic contrast administration; alert the radiology team
- Temperature extremes: very hot or cold environments, sudden temperature changes
- Alcohol: even small amounts can trigger degranulation in sensitive patients
Venom Immunotherapy
Venom immunotherapy (VIT) — subcutaneous desensitization with Hymenoptera venom extract — is strongly recommended for all SM patients who have experienced venom-triggered anaphylaxis or who have venom sensitization (positive skin test or specific IgE). Key points for SM-specific VIT:
- VIT dramatically reduces the risk of future severe anaphylaxis from Hymenoptera stings, potentially from >50% risk per sting to less than 5%
- In the general population, VIT is typically administered for 3-5 years; for SM patients, lifelong VIT is recommended because the risk of anaphylaxis recurs upon discontinuation in the setting of persistent elevated mast cell burden
- Epinephrine auto-injector should be continued even during VIT
Treatment — Advanced Systemic Mastocytosis
Advanced SM — including aggressive SM (ASM), SM with associated hematologic neoplasm (SM-AHN), and mast cell leukemia (MCL) — requires cytoreductive therapy to reduce mast cell burden and reverse organ damage.
Avapritinib (Ayvakit)
Avapritinib (Blueprint Medicines) represents the most significant therapeutic advance in mastocytosis treatment. It is a highly selective type I kinase inhibitor designed specifically to inhibit the D816V mutant form of KIT. This selectivity is critical: the KIT D816V mutation is in the activation loop of the kinase and confers resistance to imatinib and other earlier-generation tyrosine kinase inhibitors that target the inactive (type II) kinase conformation.
- FDA approval for advanced SM (2021): avapritinib 200 mg once daily for ASM, SM-AHN, and MCL; trial (EXPLORER; Search PubMed) demonstrated an overall response rate exceeding 75% in advanced SM patients; deep reductions in serum tryptase, bone marrow mast cell percentage, and spleen volume were observed
- FDA approval for indolent SM (2023): avapritinib 25 mg once daily for ISM patients with severe, refractory mediator symptoms; lower dose achieves meaningful symptom control while reducing the intracranial bleeding risk observed at higher doses
- Common side effects: periorbital edema, dizziness, cognitive effects ("avapritinib brain fog"), nausea; intracranial bleeding is a rare but serious concern, particularly in patients on anticoagulation
Midostaurin (Rydapt)
Midostaurin (Novartis) was the first FDA-approved drug for advanced SM (2017). It is a multi-kinase inhibitor targeting KIT D816V along with FLT3, PKC-alpha, PKC-beta, and VEGFR. While less KIT D816V-selective than avapritinib, it achieves partial responses in approximately 60% of advanced SM patients. Midostaurin 100 mg twice daily (with food) is the approved regimen. Common side effects include nausea, vomiting, and fatigue. It is also approved for FLT3-mutated acute myeloid leukemia, which is relevant for SM-AHN patients with concurrent AML.
Cladribine (2-CdA)
Cladribine, a purine nucleoside analog, was a principal cytoreductive agent for ASM before the KIT inhibitor era. It induces mast cell apoptosis through incorporation into DNA during replication and inhibition of DNA repair. Cladribine reduces mast cell burden and improves C-findings in a subset of patients but produces significant immunosuppression (particularly CD4+ T cell depletion). It remains an option in resource-limited settings or in patients not eligible for KIT inhibitors.
Interferon-Alpha
Interferon-alpha (with or without corticosteroids) was used in earlier decades as cytoreductive therapy for ASM. Response rates were moderate and treatment was associated with substantial toxicity (flu-like symptoms, depression, cytopenias). With the availability of avapritinib and midostaurin, interferon-alpha has largely been displaced from frontline advanced SM treatment but may still be used in certain situations.
Allogeneic Stem Cell Transplantation
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is considered for patients with MCL or refractory ASM who are medically fit for the procedure and have a suitable donor. Allo-HSCT can achieve long-term remission but carries substantial treatment-related mortality (20-30% in most series for advanced SM patients given their typically older age and organ dysfunction). The availability of effective KIT inhibitors has altered the treatment sequencing, and allo-HSCT is generally reserved for patients who fail or are intolerant to avapritinib-based therapy.
Hereditary Alpha-Tryptasemia
Hereditary alpha-tryptasemia (HαT) is a recently characterized germline condition that must be distinguished from mastocytosis in patients with elevated baseline serum tryptase and systemic symptoms.
Genetics and Mechanism
HαT is caused by increased copy numbers of the TPSAB1 gene, which encodes alpha-tryptase. Normally, individuals carry two copies of TPSAB1 (one per chromosome). In HαT, one or both chromosomes carry additional TPSAB1 copies (duplex, triplex, or tetraplex), resulting in constitutively increased tryptase production even in the absence of mast cell clonal expansion. HαT follows an autosomal dominant inheritance pattern and is present in approximately 5% of the general population.
Clinical Presentation
HαT produces a multisystem symptom complex that substantially overlaps with mastocytosis and mast cell activation syndrome:
- Elevated baseline serum tryptase (often 8-40 ng/mL), sometimes exceeding the 20 ng/mL minor SM criterion
- Flushing, urticaria, pruritus
- GI symptoms including abdominal pain and diarrhea
- Connective tissue abnormalities (joint hypermobility, which may satisfy criteria for hypermobile Ehlers-Danlos syndrome)
- Autonomic dysfunction (orthostatic intolerance, POTS-like features)
- Atopy and allergic sensitivities
Distinguishing HαT from Mastocytosis
The distinction is critical because the prognosis and management differ substantially:
- In HαT: KIT D816V mutation is absent; bone marrow biopsy does not show mast cell aggregates; WHO SM criteria are not fulfilled; no clonal mast cell disease
- In SM: KIT D816V typically present; BM biopsy shows aggregates; WHO criteria are met
- TPSAB1 copy number genotyping (available as a clinical laboratory test) confirms HαT
- HαT and SM can coexist in the same patient, and HαT may amplify the tryptase elevation of SM, making the serum tryptase threshold less specific in this context
Management of HαT focuses on symptom control (antihistamines, avoiding triggers) rather than cytoreductive therapy; no mast cell cytoreduction is indicated because there is no clonal expansion.
Key Research Papers
- Valent P et al. "Diagnostic criteria and classification of mastocytosis: a consensus proposal." Int Arch Allergy Immunol. 2014. — Search PubMed
- Pardanani A. "Systemic mastocytosis in adults: 2019 update on diagnosis, risk stratification, and management." Am J Hematol. 2019. — Search PubMed
- Gotlib J et al. "Efficacy and Safety of Avapritinib in Advanced Systemic Mastocytosis." N Engl J Med. 2021. — Search PubMed
- Radia DH, Terpos E. "Diagnosis and treatment of systemic mastocytosis." Pharmacol Ther. 2017. — Search PubMed
- Siebenhaar F et al. "Hereditary alpha tryptasemia is a common cause of elevated basal serum tryptase in patients with mastocytosis." J Allergy Clin Immunol. 2018. — Search PubMed
- Cohen SS et al. "Avapritinib in indolent systemic mastocytosis." N Engl J Med. 2023. — Search PubMed
- Brockow K et al. "Anaphylaxis in patients with mastocytosis: a study on history, clinical features and risk factors in 120 patients." Ann Allergy Asthma Immunol. 2014. — Search PubMed
- van Anrooij B et al. "Serum tryptase in the diagnosis of systemic mastocytosis: a prospective study." Clin Chem Lab Med. 2018. — Search PubMed
- Rossini M et al. "Bone density and skeletal complications of mastocytosis: a multicenter study." Osteoporos Int. 2014. — Search PubMed
- Zanotti R et al. "Osteoporosis in systemic mastocytosis and other rare hematological malignancies." Haematologica. 2015. — Search PubMed
- Carter MC et al. "Pediatric mastocytosis: routine anesthetic management for a complex disease." J Allergy Clin Immunol. 2007. — Search PubMed
- Akin C. "Mast cell activation syndromes." J Allergy Clin Immunol. 2017. — Search PubMed
PubMed Topic Searches
- Systemic mastocytosis diagnosis and treatment — PubMed
- KIT D816V mutation mast cell — PubMed
- Avapritinib mastocytosis — PubMed
- Mastocytosis anaphylaxis venom immunotherapy — PubMed
- Hereditary alpha-tryptasemia TPSAB1 — PubMed
Connections
- Pain & Allergy
- Histamine, Mast Cells & Allergy — interactive animation
- Mast Cell Activation Syndrome (MCAS)
- Allergies
- Anaphylaxis
- Hereditary Angioedema
- Common Variable Immunodeficiency
- MCAS: History and Discovery
- Chronic Granulomatous Disease
- Tryptase — serum total tryptase is the primary biomarker used to screen for and monitor mastocytosis.