Becker Muscular Dystrophy (BMD)
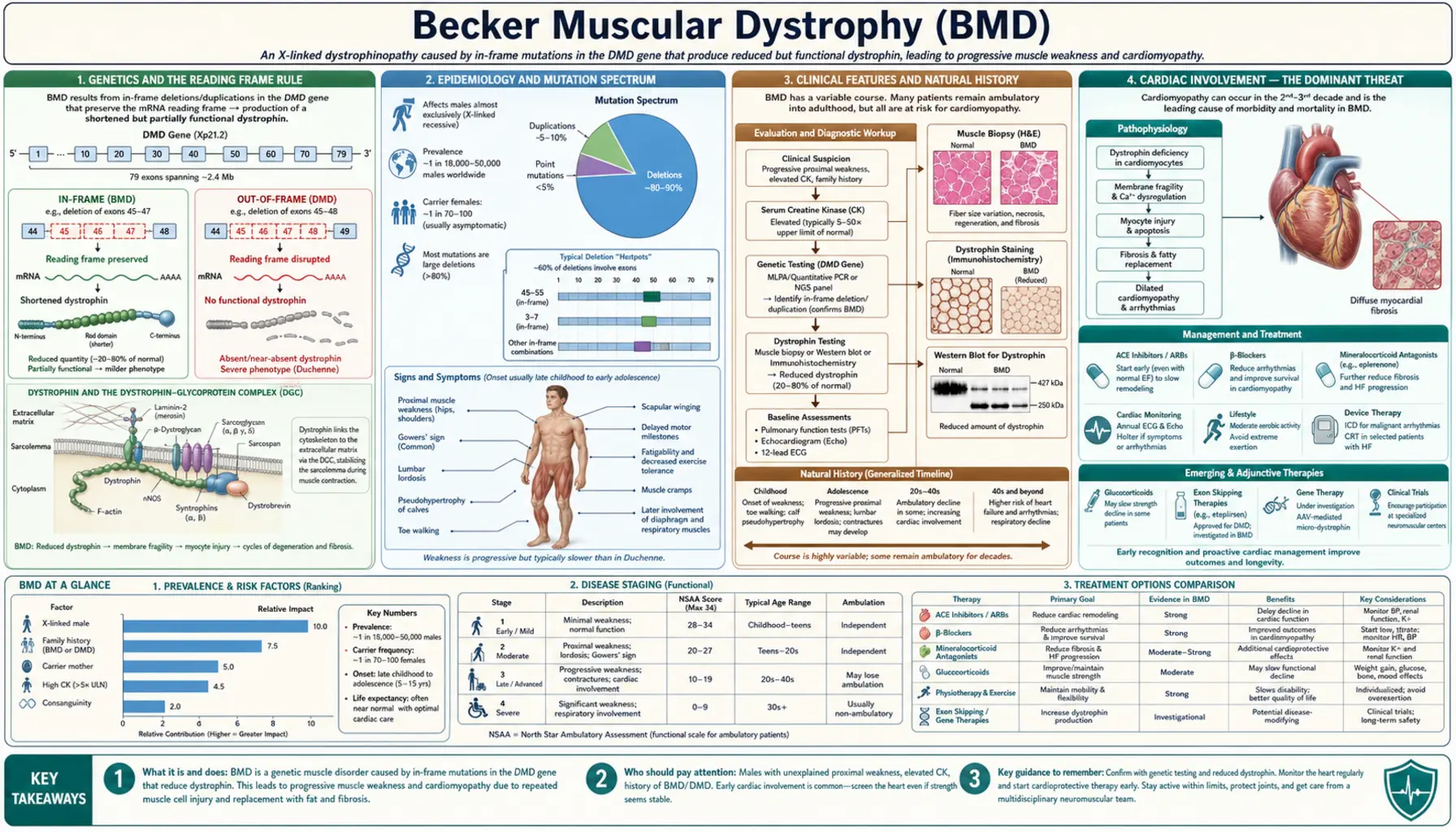
Becker muscular dystrophy (BMD) is an X-linked recessive neuromuscular disease caused by in-frame mutations in the DMD gene (Xp21.2) that allow production of a truncated but partially functional dystrophin protein. Because some dystrophin reaches the sarcolemma and stabilizes the dystrophin-glycoprotein complex, the phenotype is significantly milder than Duchenne muscular dystrophy — with onset typically in adolescence or early adulthood and ambulation often preserved into the fifth or sixth decade. However, dilated cardiomyopathy in BMD is frequently more severe relative to skeletal muscle involvement and can dominate the clinical picture, making cardiac surveillance a cornerstone of management.
Interactive Visualization Muscle Contraction — release the calcium Expose the actin binding sites and watch myosin heads ratchet through the cross-bridge cycle to shorten the sarcomere. Launch →Table of Contents
- Genetics and the Reading Frame Rule
- Epidemiology and Mutation Spectrum
- Clinical Features and Natural History
- Cardiac Involvement — The Dominant Threat
- Quadriceps Myopathy and Exercise Intolerance
- Diagnosis
- Management
- Key Research Papers
- Featured Videos
Genetics and the Reading Frame Rule
Becker muscular dystrophy and Duchenne muscular dystrophy both arise from mutations in the same gene — the DMD gene located at Xp21.2. At 2.4 megabases and 79 exons, the DMD gene is the largest known human gene. The disorder follows an X-linked recessive inheritance pattern, meaning that males with a single mutant allele are affected and females carrying one mutant allele are generally carriers, though some develop symptoms.
The fundamental distinction between BMD and DMD was explained by the Monaco Reading Frame Rule, published in 1988. In-frame deletions or duplications — those that preserve the translational reading frame — allow the ribosome to continue reading the mRNA and produce a dystrophin protein that, while internally deleted or shortened, retains partial function. These truncated proteins can still partially anchor the dystrophin-glycoprotein complex at the muscle cell membrane, producing the milder BMD phenotype. Out-of-frame mutations, by contrast, introduce a premature stop codon downstream, resulting in no stable dystrophin protein and the severe DMD phenotype.
The most common BMD mutations are large in-frame exon deletions, particularly those spanning exons 45–47 or 45–51, a region often called the "BMD hotspot." Some duplications also produce in-frame transcripts. Rare point mutations (missense or splice-site) can also yield partially functional protein. On Western blot, BMD muscle shows dystrophin at reduced quantity and abnormal molecular weight compared with normal, in contrast to the virtual absence seen in DMD. Patients with dystrophin levels of 10–40% of normal tend toward very mild phenotypes, while those with 3–10% overlap the severe BMD range.
Genotype-phenotype correlation is imperfect — the same exon 45–51 deletion can produce ambulatory patients in their sixties in one family and early wheelchair dependence in another. Modifier genes including SPP1 (osteopontin) and LTBP4 are under active investigation as contributors to this variability. Brain involvement is uncommon in BMD because the shorter Dp140 and Dp71 dystrophin isoforms, which are expressed in the brain and are disrupted in most DMD deletions, are usually at least partially preserved by in-frame BMD mutations.
Epidemiology and Mutation Spectrum
BMD is considerably less common than DMD. Prevalence estimates range from approximately 1 in 18,000 to 1 in 31,000 live male births, translating to roughly 5,000–6,000 patients in the United States at any given time. The disease occurs worldwide without clear ethnic predisposition.
Approximately 80% of BMD cases result from in-frame exon deletions, 10% from duplications, and 10% from point mutations (missense, nonsense, or splice-site changes that nonetheless allow some functional dystrophin). The single most common mutation — an in-frame deletion of exons 45–51 — accounts for approximately 30% of all BMD cases and is associated with a particularly mild phenotype: many affected males remain ambulatory well into their sixties and seventies.
Approximately 30% of BMD cases arise from de novo mutations with no prior family history, a figure similar to DMD. Genetic counseling is therefore important even when no family history is apparent. Female carriers of DMD gene mutations have an approximately 8% lifetime risk of developing dilated cardiomyopathy themselves, even when skeletal muscle function is entirely normal. For this reason, cardiac screening is recommended for all female carriers regardless of symptoms.
A clinically important subset of patients with DMD gene mutations present as X-linked dilated cardiomyopathy (XLDCM) — a condition characterized by isolated or predominant cardiomyopathy with minimal or absent skeletal muscle weakness. XLDCM is typically caused by mutations that selectively affect the muscle-specific promoter region or exon 1 of the DMD gene, impairing cardiac dystrophin expression while leaving enough skeletal muscle isoform function intact to prevent obvious weakness. Young males presenting with unexplained dilated cardiomyopathy should always have DMD gene testing included in the workup.
Clinical Features and Natural History
BMD typically presents between ages 5 and 25, in contrast to DMD where weakness is usually evident by ages 2–5. The range is wide — some mildly affected males are not diagnosed until they present with exertional myoglobinuria or cardiomyopathy in their thirties or forties. Others have a more severe course that resembles late-onset DMD.
The distribution of weakness mirrors DMD — proximal muscles of the hip girdle and proximal lower extremities are affected first. Patients commonly notice difficulty rising from the floor (requiring a Gower maneuver in some), climbing stairs, or running. The pace of progression is substantially slower than DMD, and ambulation is typically preserved into the forties or sixties in most BMD patients. Approximately 30% require wheelchair assistance by age 40–50; the remainder may remain ambulatory much longer.
Selective quadriceps wasting with relative preservation of hip abductors is a characteristic pattern in BMD that differs subtly from DMD and can be a useful clinical clue. Calf hypertrophy, caused by fibrofatty replacement of degenerating muscle fibers, is present in many BMD patients as in DMD. Gait abnormalities — waddling, toe-walking, and a Trendelenburg pattern — are milder than in DMD. Contractures develop later and are less severe; clinically significant scoliosis is uncommon.
Cognitive involvement is rare in BMD. Unlike DMD, where disruption of the short Dp71 isoform expressed widely in the brain contributes to a higher rate of intellectual disability and attention problems, most BMD patients have entirely normal intelligence. Creatine kinase (CK) is markedly elevated, typically 5,000–20,000 IU/L in younger patients; the level may paradoxically decline in adulthood as progressive muscle wasting reduces the total mass of muscle available to leak the enzyme. Exertional rhabdomyolysis — episodes of acute muscle breakdown triggered by unaccustomed intense exercise or febrile illness — is a common and sometimes dramatic presenting feature in mildly affected adult males, producing dark cola-colored urine (myoglobinuria) and sharply elevated CK.
Cardiac Involvement — The Dominant Threat
Dilated cardiomyopathy develops in 70–80% of BMD patients by age 40, making it one of the most clinically significant features of the disease and a major driver of morbidity and mortality. Importantly, cardiac involvement in BMD does not track closely with skeletal muscle disease severity. A patient who is ambulatory and functionally independent may simultaneously harbor severe left ventricular dysfunction — a phenomenon called the cardiomyopathy-skeletal mismatch — that makes BMD cardiac disease potentially more dangerous per unit of skeletal disability than DMD, where respiratory and orthopedic complications often dominate before cardiac disease becomes the limiting factor.
The cardiac pathology in BMD is the same as in DMD: left ventricular dilatation, global systolic dysfunction with reduced ejection fraction, and progressive diastolic dysfunction. Cardiac MRI characteristically demonstrates posterolateral LV wall fibrosis on late gadolinium enhancement sequences, reflecting replacement of dystrophin-deficient cardiomyocytes by scar tissue. This fibrosis pattern may appear earlier in BMD relative to the degree of skeletal involvement. Electrocardiographic findings mirror DMD — a tall R-wave in lead V1 and deep Q-waves in inferolateral leads are characteristic.
X-linked dilated cardiomyopathy represents the extreme of cardiac-dominant BMD: affected males may have entirely normal or minimally elevated CK and no skeletal weakness, presenting to cardiology with unexplained DCM in their twenties or thirties. Without awareness of this entity, the underlying genetic cause and appropriate family counseling may be missed for years.
Cardiac screening guidelines recommend ECG and echocardiography beginning at age 10 (or at diagnosis if older), repeated annually. Cardiac MRI every 2–3 years provides superior characterization of fibrosis burden and early LV dysfunction not yet apparent on echo. All symptomatic female carriers should also undergo cardiac evaluation, and many guidelines now recommend baseline cardiac assessment for all carriers regardless of symptoms.
Treatment follows heart failure guidelines with disease-specific modifications. ACE inhibitors (enalapril, lisinopril) and beta-blockers (carvedilol, metoprolol succinate) are started early — often even before the ejection fraction falls below the normal range — because early intervention has been shown to slow progression in DMD and is assumed to apply to BMD. Mineralocorticoid receptor antagonists such as eplerenone add further cardioprotection. Patients with sustained ventricular tachycardia or ejection fraction persistently below 35% are candidates for implantable cardioverter-defibrillator placement. For ambulatory BMD patients with end-stage cardiomyopathy who retain adequate skeletal muscle function, cardiac transplantation is a legitimate and increasingly utilized option, with outcomes comparable to other transplant indications.
Quadriceps Myopathy and Exercise Intolerance
Selective quadriceps wasting is one of the clinical hallmarks that distinguishes BMD from other muscular dystrophies. Affected patients develop progressive weakness and visible atrophy of the anterior thigh muscles — particularly the rectus femoris and vastus lateralis — while other proximal muscle groups, including the hip abductors, may be relatively spared early in the disease. Patients notice this as difficulty climbing stairs, rising from a low chair or squat, and instability on uneven terrain well before global gait disturbance develops. The selective pattern can mimic an isolated quadriceps myopathy, and BMD should be in the differential for any male presenting with this pattern.
Exertional rhabdomyolysis and myoglobinuria are particularly common in mildly affected BMD adults and in some patients represent the initial presentation before a diagnosis of BMD is established. Episodes are typically triggered by unusually strenuous or unaccustomed exercise — a single intense workout, a long hike, or a competitive sport — or by febrile illness. During an episode, CK may spike above 100,000 IU/L and the urine turns dark brown (cola or tea colored) from myoglobin. The primary complication is acute kidney injury from myoglobin-induced tubular toxicity, requiring aggressive IV hydration and monitoring of renal function. Patients should carry a medical alert identification noting their diagnosis and the anesthetic contraindications.
Succinylcholine is absolutely contraindicated in BMD, as in all muscular dystrophies. The depolarizing neuromuscular blocking agent can trigger life-threatening hyperkalemia and rhabdomyolysis in these patients. Volatile inhalational anesthetic agents carry a similar risk. Anesthesiologists must be informed of the diagnosis before any surgical procedure, and a preoperative CK measurement provides a useful baseline. Non-depolarizing agents and total intravenous anesthesia are preferred.
Physical therapy guidance in BMD aims to maintain function while avoiding the muscle damage of high-intensity eccentric exercise. Moderate-intensity aerobic exercise (swimming, cycling, walking) is safe and beneficial. Resistance training at approximately 60% of maximum voluntary contraction is supported by evidence in related conditions and is generally recommended. Aquatic therapy reduces joint stress and is well tolerated. High-intensity plyometric or heavy eccentric loading should be avoided. Occupational therapy assessment helps preserve independence in daily activities as strength declines.
Diagnosis
The diagnostic workup for suspected BMD begins with serum CK measurement. CK is elevated in virtually all affected males — typically 5,000–20,000 IU/L in younger patients — and an unexpectedly high CK in an otherwise healthy-appearing male should prompt genetic evaluation. In adults with advanced muscle wasting, CK may be lower due to reduced total muscle mass; a normal CK in a middle-aged male with known family history does not exclude the diagnosis.
Genetic testing has become the definitive first-line diagnostic modality. Multiplex ligation-dependent probe amplification (MLPA) detects exon deletions and duplications across the entire DMD gene and identifies approximately 80–90% of pathogenic variants. For patients with a negative MLPA, full sequencing of the DMD gene identifies the remaining point mutations and small indels. Once a deletion or duplication is found, reading frame analysis — determining whether the mutation is in-frame or out-of-frame — is essential to predict and understand the phenotype. The same exon deletion can be BMD in one patient and DMD in another depending on the precise boundaries and reading frame consequences.
Muscle biopsy is reserved for patients in whom genetic testing is inconclusive or when the diagnosis remains in question. Histology in BMD shows the hallmarks of a dystrophic myopathy — fiber size variation, internal nuclei, necrosis and regeneration — but these changes are less severe than in DMD. The critical finding is dystrophin immunostaining: in BMD, dystrophin is present but reduced in intensity and sometimes patchy in distribution, in contrast to DMD where staining is absent or nearly absent. Western blot demonstrates reduced quantity and abnormal (lower) molecular weight of the dystrophin protein band, directly reflecting the internally deleted protein.
Cardiac evaluation — ECG and echocardiography — should be performed at diagnosis in all patients regardless of age, because cardiac disease may already be present or may progress independently of skeletal muscle disease. Electromyography reveals a myopathic pattern (short-duration, low-amplitude, polyphasic motor unit potentials) and normal nerve conduction studies, helping exclude neuropathic causes of weakness such as Charcot-Marie-Tooth disease or spinal muscular atrophy. The differential diagnosis includes other limb-girdle muscular dystrophies (particularly the sarcoglycanopathies, LGMD2C-2F, where dystrophin is normal), XLDCM without skeletal weakness, sporadic dilated cardiomyopathy, inflammatory myopathies such as polymyositis (which typically show elevated inflammatory markers and respond to steroids), and mitochondrial myopathy.
Management
BMD management is multidisciplinary, encompassing neurology, cardiology, physical medicine and rehabilitation, genetic counseling, and psychosocial support. The goal is to preserve function, prevent complications, and maintain quality of life across what may be a decades-long disease course.
No exon-skipping therapies are specifically approved for BMD by the FDA. The exon-skipping drugs approved for specific DMD genotypes (eteplirsen for exon 51 skipping, golodirsen and viltolarsen for exon 53 skipping, casimersen for exon 45 skipping) theoretically apply to BMD patients whose mutations could be shifted further out of frame by additional exon deletion — a concept of "exon-skipping to worsen the reading frame" that would paradoxically improve phenotype. This remains an area of active clinical investigation. The recently approved micro-dystrophin gene therapy (Elevidys, delandistrogene moxeparvovec) includes BMD patients in ongoing extension trials.
Corticosteroids (prednisone or deflazacort) have a well-established role in slowing progression in DMD but evidence in BMD is more limited. For patients with a severe or intermediate phenotype — particularly those who are losing ambulation — a trial of corticosteroids at individualized dosing is a reasonable consideration after shared decision-making about side effects. Typical dosing extrapolated from DMD protocols (prednisone 0.75 mg/kg/day or deflazacort 0.9 mg/kg/day) is used; weekend-only regimens reduce but do not eliminate side effects.
Cardiac management is the highest-priority therapeutic domain given the frequency and severity of BMD cardiomyopathy. Early initiation of ACE inhibitors and beta-blockers — even when ejection fraction remains preserved — is supported by evidence from DMD studies and expert consensus. Diuretics manage fluid overload. Aldosterone antagonists (eplerenone or spironolactone) provide additive cardioprotection. Patients with ejection fraction below 35% or life-threatening arrhythmia are referred for ICD implantation. Ambulatory BMD patients with end-stage cardiomyopathy who retain reasonable skeletal muscle function are appropriate candidates for cardiac transplantation; this is increasingly recognized as a viable and appropriate intervention.
Physical and occupational therapy should be scheduled at regular intervals — every 6–12 months — to monitor functional status, adjust orthotic devices, and update exercise prescriptions. Ankle-foot orthoses help manage foot drop when it develops. Surgical correction of contractures is rarely necessary given the slower progression compared with DMD. Scoliosis surveillance is appropriate but surgical intervention is uncommon. Psychosocial screening for depression and anxiety is important; the late-onset, slowly progressive nature of BMD means many patients have established careers, relationships, and independent lives before significant disability develops, and the progressive course requires ongoing psychological adjustment.
Genetic counseling is essential for the patient and family. The X-linked pattern means that affected males pass the mutation to all daughters (who become carriers) but to none of their sons. Carrier females have a 50% chance of having an affected son and a 50% chance of having a carrier daughter with each pregnancy. Preimplantation genetic testing and prenatal diagnosis are available options. All female relatives in the maternal line should be offered carrier testing and, if confirmed, cardiac evaluation.
Key Research Papers
- Monaco AP et al. "An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus." Genomics. 1988;2(1):90–5. — Search PubMed
- Hoffman EP et al. "Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy." N Engl J Med. 1988;318(21):1363–8. — Search PubMed
- Nigro V et al. "Identification of the Becker muscular dystrophy gene product." Hum Mol Genet. 1994;3(9):1597–600. PMID: 7833920
- Melacini P et al. "Myocardial involvement is very frequent among patients affected with subclinical Becker's muscular dystrophy." Circulation. 1996;94(12):3168–75. — Search PubMed
- Muntoni F et al. "Deficiency of dystrophin associated proteins in Duchenne muscular dystrophy patients." Lancet. 1995;345(8942):91–2. — Search PubMed
- Finsterer J et al. "Cardiac involvement in Becker muscular dystrophy." Can J Cardiol. 2008;24(10):786–92. — Search PubMed
- Bushby KM et al. "The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy." J Neurol. 1993;240(2):98–104. — Search PubMed
- Manzur AY et al. "Glucocorticoid corticosteroids for Duchenne muscular dystrophy." Cochrane Database Syst Rev. 2008;(1):CD003725. — Search PubMed
- Politano L et al. "Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies." JAMA. 1996;275(17):1335–8. — Search PubMed
- Quinlivan R et al. "An association analysis of Duchenne muscular dystrophy modifier genes." Brain. 2010;133(Pt 8):2316–26. — Search PubMed
- Emery AE. "The muscular dystrophies." Lancet. 2002;359(9307):687–95. PMID: 11879882
- McNally EM et al. "Contemporary cardiac issues in Duchenne muscular dystrophy." Circulation. 2015;131(18):1590–8. — Search PubMed
Connections
- Neurology
- Muscle Contraction — interactive animation
- Duchenne Muscular Dystrophy (DMD)
- Facioscapulohumeral Muscular Dystrophy
- Myotonic Dystrophy
- Spinal Muscular Atrophy
- ALS
- Myasthenia Gravis
- Lab Tests