ALS: History and Discovery
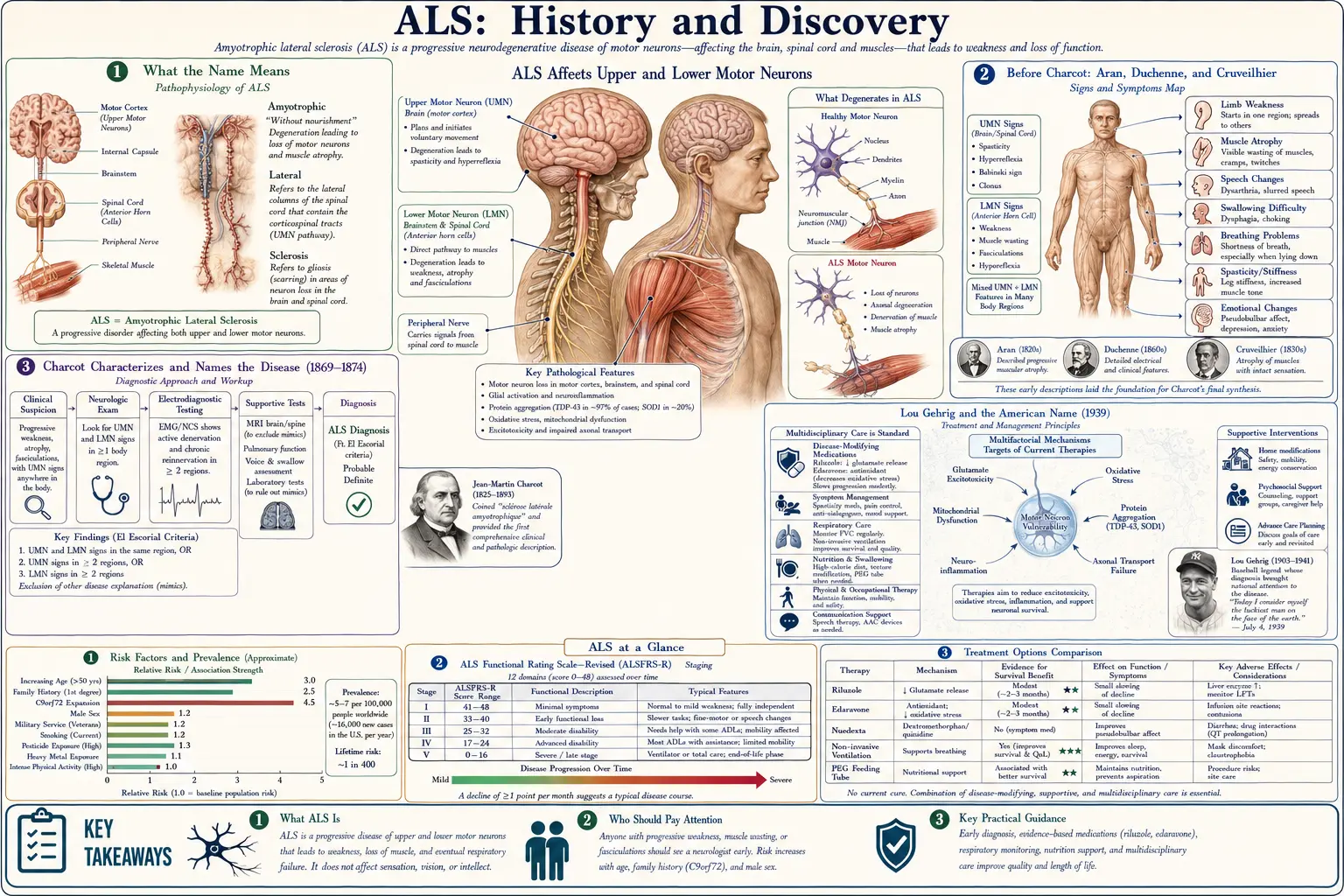
Amyotrophic lateral sclerosis (ALS) was named by the French neurologist Jean-Martin Charcot at the Salpêtrière hospital in Paris, who described its clinical picture in 1869 and coined the term sclérose latérale amyotrophique in 1874 — correlating progressive paralysis and wasting with the hardening (sclerosis) of the lateral corticospinal tracts and the loss of the anterior horn motor neurons. In France it is still called Charcot's disease; in the United States it is known as Lou Gehrig's disease after the New York Yankees first baseman diagnosed in 1939. Charcot built on earlier descriptions of muscular wasting by Aran, Duchenne, and Cruveilhier rather than discovering the condition from nothing. The first ALS gene, SOD1, was identified in 1993, and the commonest genetic cause, the C9orf72 repeat expansion, in 2011 — opening the molecular era that produced the first gene-targeted ALS drug in 2023.
Table of Contents
- What the Name Means
- Before Charcot: Aran, Duchenne, and Cruveilhier
- Charcot Characterizes and Names the Disease (1869–1874)
- Lou Gehrig and the American Name (1939)
- The Twentieth Century: Spectrum, Criteria, and Clues
- SOD1 and the First ALS Gene (1993)
- TDP-43 and C9orf72: The Modern Molecular Era (2006–2011)
- From Riluzole to Gene-Targeted Therapy
- Legacy, Advocacy, and Open Questions
- Research Papers and References
- Connections
- Featured Videos
What the Name Means
The full name amyotrophic lateral sclerosis is a precise, two-part clinical-pathological description, and unpacking it explains almost everything about the disease. Amyotrophic comes from the Greek — a- (no), myo- (muscle), trophic (nourishment): "no muscle nourishment," the wasting and weakness that follow when the lower motor neurons of the anterior horn of the spinal cord die and the muscles they supply lose their nerve signal. Lateral sclerosis refers to the hardening (scarring, or "sclerosis") that Charcot saw in the lateral columns of the spinal cord — the descending corticospinal tracts that carry commands from the brain's upper motor neurons. The name therefore encodes the disease's defining feature: it strikes both the upper motor neurons (causing spasticity and brisk reflexes) and the lower motor neurons (causing wasting, weakness, and muscle twitching, or fasciculation).
This dual-pathology insight was Charcot's great contribution. Earlier physicians had described the muscle-wasting half of the picture and assumed it was a primary disease of the muscle; Charcot recognized that the muscle atrophy and the spinal-cord hardening were two visible faces of a single disorder of the motor system, and he chose a name that captured both at once. The term has proved remarkably durable: more than 150 years later it remains the standard medical name worldwide, alongside the two great eponyms the disease has acquired — which are geographic.
In France and much of continental Europe, ALS is Charcot's disease (maladie de Charcot), honoring the man who defined it. In the United States — and increasingly in popular usage elsewhere — it is Lou Gehrig's disease, after the baseball star whose 1939 diagnosis brought the condition into millions of homes. In the United Kingdom and parts of the Commonwealth the broader umbrella term motor neurone disease (MND) is often preferred, with ALS understood as its most common form. One disease, several names, each a window onto a different chapter of its history.
Before Charcot: Aran, Duchenne, and Cruveilhier
Charcot is rightly credited with characterizing and naming ALS, but he did not describe motor-neuron disease from nothing — he synthesized and surpassed the work of predecessors, and honesty about that lineage matters. The earliest threads reach back to the Scottish anatomist Sir Charles Bell, who in the 1820s and 1830s helped establish that the anterior (ventral) spinal roots carry motor function while the posterior roots carry sensation, the anatomical foundation on which any understanding of a pure motor disease must rest. Scattered case reports of progressive limb wasting appear in the early nineteenth-century literature, but they lacked a unifying framework.
The decisive pre-Charcot figure was the Parisian physician François-Amilcar Aran, who in 1850 published a series of eleven patients with progressive weakness and wasting of the limbs and named the condition progressive muscular atrophy (PMA). Aran had compiled and studied these cases with the help of Guillaume-Benjamin-Amand Duchenne de Boulogne, the pioneering clinical neurologist; Duchenne later claimed to have recognized the disorder first, and the condition was for a time called Aran–Duchenne disease (or Duchenne–Aran), a genuine priority dispute that historians still note. Crucially, both Aran and Duchenne initially believed PMA was a disease of muscle itself.
That assumption was overturned by the great pathologist Jean Cruveilhier, who in 1853 examined the autopsy of one of Aran's patients and found atrophy of the anterior spinal roots and the motor nerves — the first solid evidence that the muscle wasting was neurogenic, arising from the nervous system rather than the muscle. Cruveilhier thus located the lesion where it belongs, in the motor pathways. These three contributions — Bell's motor-root anatomy, Aran's clinical syndrome, and Cruveilhier's neuropathology — were the materials Charcot inherited. His achievement was to gather them, add the upper-motor-neuron (lateral tract) dimension, and forge a single, named disease entity.
Charcot Characterizes and Names the Disease (1869–1874)
Jean-Martin Charcot (1825–1893), working at the Hôpital de la Salpêtrière in Paris — a vast institution often called the cradle of modern neurology — turned the disorder into a defined disease through his signature méthode anatomo-clinique (the anatomo-clinical method): meticulously documenting a living patient's signs and symptoms, then correlating them, after death, with the precise lesions found at autopsy. With his collaborator Alix Joffroy, Charcot studied cases in which paralysis with spasticity (but little wasting) tracked to lesions of the spinal cord's lateral columns, while paralysis with wasting tracked to disease of the anterior horn cells. He saw that ALS combined both.
In 1869 Charcot reported the first cases, in a paper on progressive muscular atrophy with lesions of the spinal cord's grey matter and anterolateral columns — the clinical-pathological correlation that defined the new entity. He continued lecturing on it in the early 1870s, and it was in 1874, as his Salpêtrière lectures were gathered and published, that he gave the disease its enduring name: sclérose latérale amyotrophique — amyotrophic lateral sclerosis. The two dates matter and are sometimes conflated: 1869 marks the characterization, 1874 marks the naming.
Charcot used ALS as a showcase for the power of his method — a disease whose clinical signs could be read backward to predict the exact anatomical damage — which is why it bears his name in France and why he is remembered as a founder of neurology. It is worth being precise about credit, however: Charcot did not discover the cause of ALS (still incompletely understood today), and he was not the first to describe progressive muscular wasting. What he did was define ALS as a distinct clinical-pathological disease and name it — an act of synthesis and classification that has structured every subsequent investigation.
Lou Gehrig and the American Name (1939)
If Charcot gave ALS its scientific identity, an American baseball player gave it a human face. Henry Louis "Lou" Gehrig, the New York Yankees' durable first baseman nicknamed "the Iron Horse" for his record streak of 2,130 consecutive games, began to falter inexplicably during the 1938–1939 seasons — losing strength, coordination, and power at the plate. After a battery of tests at the Mayo Clinic in Rochester, Minnesota, doctors confirmed the diagnosis of amyotrophic lateral sclerosis on June 19, 1939 — Gehrig's 36th birthday. The prognosis was blunt: progressive paralysis and a life expectancy of only a few years, though his mind would remain clear.
On July 4, 1939, the Yankees held "Lou Gehrig Appreciation Day" at Yankee Stadium before a crowd reported at 61,808. There Gehrig delivered what became one of the most famous speeches in American sports, telling the hushed stadium, "Today I consider myself the luckiest man on the face of the earth" — a moment of grace from a dying man that fixed both the player and his disease permanently in the public memory. His former teammate Babe Ruth embraced him as the ceremony ended. Gehrig died less than two years later, on June 2, 1941, at the age of 37.
Because Gehrig was a national figure and his decline so public, Americans began calling ALS "Lou Gehrig's disease," and the eponym stuck so firmly that for decades many people in the United States knew the condition by no other name. It was the most consequential public-awareness event in the disease's history until the twenty-first century. (A historical footnote worth flagging honestly: a small number of later researchers have speculated that the repeated head trauma of contact sports can cause an ALS-like syndrome, raising questions about Gehrig's own case; this remains unproven speculation, and the established record is that Gehrig was diagnosed with and died of ALS.)
The Twentieth Century: Spectrum, Criteria, and Clues
For nearly a century after Charcot, ALS remained clinically recognizable but mechanistically opaque — a relentless dying-back of motor neurons with no known cause and no treatment. Twentieth-century neurology refined the picture in three main ways. First, clinicians came to understand ALS as part of a wider motor neurone disease spectrum that also includes progressive muscular atrophy (lower-motor-neuron predominant), primary lateral sclerosis (upper-motor-neuron predominant), and progressive bulbar palsy (beginning in the muscles of speech and swallowing), with classic ALS combining upper and lower signs.
Second, epidemiologists noticed striking geographic clusters — most famously the very high incidence of an ALS–parkinsonism–dementia complex among the Chamorro people of Guam in the mid-twentieth century. Decades of investigation linked it provisionally to environmental factors, including a neurotoxin (BMAA) associated with traditional cycad-seed consumption, though no single cause has ever been definitively proven and the cluster has since declined. The Guam puzzle nonetheless kept alive the idea that ALS might have environmental as well as genetic triggers.
Third, to make research comparable across centers, neurologists developed formal diagnostic criteria — most influentially the El Escorial criteria, issued by the World Federation of Neurology in 1994 and revised thereafter — which grade diagnostic certainty by how many body regions show combined upper- and lower-motor-neuron signs. These criteria standardized clinical-trial enrollment even as the underlying biology stayed mysterious. The mystery began to break only when genetics arrived.
SOD1 and the First ALS Gene (1993)
The molecular era of ALS opened in 1993, when Daniel Rosen and colleagues, reporting in Nature, identified mutations in the gene encoding the enzyme copper/zinc superoxide dismutase 1 (SOD1) in families with inherited ALS. This was the first ALS gene ever discovered, and it was a landmark: for the first time, a concrete molecular defect could be tied to motor-neuron death. SOD1 mutations account for a substantial minority of familial (inherited) ALS — commonly cited at roughly 12–20% — though familial cases themselves make up only about 10% of all ALS, the remaining ~90% being "sporadic" with no clear family history.
The discovery was scientifically surprising. SOD1 is an antioxidant enzyme that defends cells against free radicals, so the obvious hypothesis was that mutations crippled its protective function and let oxidative damage kill neurons. Years of work, however, pointed instead to a toxic gain of function: the mutant protein misfolds and aggregates, harming motor neurons through new toxic properties rather than simply through loss of antioxidant activity. (This distinction — loss-of-function versus gain-of-function — is presented here as the prevailing interpretation; the precise mechanism has been debated for three decades.) The SOD1 mouse, engineered to carry a mutant human gene, became the first animal model of ALS and the workhorse of preclinical research.
SOD1's importance extends beyond its share of cases: it established the template for ALS gene-hunting, created the first model system for testing therapies, and — thirty years later — became the target of the first gene-directed ALS drug (see below). The 1993 finding did not explain sporadic ALS, but it proved that ALS was, at least in part, a genetic disease and gave researchers their first molecular foothold.
TDP-43 and C9orf72: The Modern Molecular Era (2006–2011)
Two discoveries in the late 2000s and early 2010s reshaped the understanding of ALS more than anything since Charcot. The first came in 2006, when Manuela Neumann and colleagues, writing in Science, identified the protein TDP-43 (TAR DNA-binding protein 43) as the major component of the abnormal, ubiquitin-tagged protein clumps found in the dying neurons of most ALS patients — including the great majority with sporadic disease — and also in many cases of frontotemporal dementia (FTD). This was pivotal because it gave the common, non-SOD1 form of ALS a shared molecular signature and revealed a deep biological link between ALS and FTD, two conditions that clinicians had increasingly recognized overlap. Today ALS is widely viewed as, in most cases, a TDP-43 proteinopathy.
The second breakthrough came in 2011, when two research groups — one led by Mariely DeJesus-Hernandez and one by Alan Renton, both publishing in Neuron — independently identified an expanded GGGGCC hexanucleotide repeat in the C9orf72 gene on chromosome 9p21 as a cause of ALS and FTD. This turned out to be the most common known genetic cause of ALS (and of FTD), far exceeding SOD1, and it accounts for a large share of familial cases and a meaningful fraction of apparently sporadic ones. The C9orf72 expansion cemented the ALS–FTD connection at the genetic level and gave the field its single most important risk gene.
Alongside SOD1, TDP-43, and C9orf72, researchers have identified mutations in other genes — including FUS, TARDBP (which encodes TDP-43 itself), and many more — so that ALS is now understood as genetically heterogeneous, with converging themes of protein misfolding and aggregation, disordered RNA processing, and failures of cellular waste-clearance. These molecular insights did not arrive in Charcot's clinical-pathological language, but they are the direct descendants of his core idea: read the disease backward from its lesions to its cause. The lesions are now molecular.
From Riluzole to Gene-Targeted Therapy
For most of ALS history there was no drug that altered the disease at all — care was supportive. That changed, modestly, in 1995, when the U.S. Food and Drug Administration approved riluzole (brand name Rilutek), the first medication shown to slow ALS and extend survival, thought to act by reducing glutamate-driven excitotoxic damage to motor neurons. Its benefit is real but limited — on the order of a few months of added survival — and for more than two decades it stood alone as the only systemic ALS therapy.
A second drug arrived in 2017, when the FDA approved edaravone (Radicava), an antioxidant free-radical scavenger, as the first new ALS treatment in 22 years; it can slow functional decline in a subset of patients. The most historically significant advance, however, came on April 25, 2023, when the FDA granted accelerated approval to tofersen (Qalsody), an antisense oligonucleotide that lowers production of the SOD1 protein and is the first therapy to target a genetic cause of ALS — closing a thirty-year loop back to the 1993 discovery of the SOD1 gene. It is approved specifically for the small population of patients carrying SOD1 mutations.
The honest picture for patients today is one of incremental but accelerating progress: ALS remains incurable and ultimately fatal, and current drugs slow rather than stop it. But the arrival of a gene-targeted therapy, the explosion of genetic and biomarker knowledge (such as neurofilament light chain as a marker of nerve injury), and a deep pipeline of antisense and other approaches mark the most hopeful era in the disease's long history — a trajectory running straight from Charcot's autopsy table to a precision drug aimed at a single gene.
Legacy, Advocacy, and Open Questions
ALS occupies an outsized place in public consciousness relative to its incidence (roughly 1–2 new cases per 100,000 people per year), in large part because of the people who have carried it into view. Lou Gehrig made it a household name in 1939. The physicist Stephen Hawking, who lived with a very slowly progressive form of ALS for more than five decades after his early-1960s diagnosis, became a global symbol of the mind's endurance against the body's failure — while also illustrating, honestly, how unusually variable the disease's course can be (most patients survive only a few years; Hawking was a rare exception, not the rule).
In 2014, the viral Ice Bucket Challenge raised well over $100 million for ALS research worldwide in a single summer — an unprecedented surge of grassroots funding that helped accelerate gene discovery and clinical trials. This blend of celebrity, patient advocacy, and mass philanthropy has shaped the modern research landscape as much as any laboratory finding, and it reflects the same human dimension that runs from Charcot's patients at the Salpêtrière to Gehrig's farewell at Yankee Stadium.
The central scientific question Charcot could not answer in 1874 remains only partly answered in our own time: what makes motor neurons die? Genetics has supplied powerful pieces — misfolded SOD1, aggregated TDP-43, the C9orf72 repeat — and has explained the inherited minority of cases, but the cause of the sporadic majority is still incompletely understood, likely a convergence of genetic susceptibility and environmental and age-related factors. The history of ALS is thus an unfinished story: a disease named more than 150 years ago, illuminated dramatically in the last three decades, and still awaiting the discovery that turns a slowed disease into a stopped one.
Research Papers and References
The references below combine landmark peer-reviewed papers in the history and molecular biology of ALS with curated PubMed topic-search links and authoritative patient resources. Where a stable identifier (DOI or PMID) is given for a specific historical or landmark paper, it has been verified; broader topics are provided as PubMed searches. Each link opens in a new tab.
- Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59-62. — PubMed PMID: 8446170
- Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130-133. — doi:10.1126/science.1134108
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245-256. — doi:10.1016/j.neuron.2011.09.011
- Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257-268. — PubMed PMID: 21944779
- Goetz CG. Amyotrophic lateral sclerosis: early contributions of Jean-Martin Charcot. Muscle & Nerve. 2000;23(3):336-343. — PubMed PMID: 10679709
- Rowland LP. How amyotrophic lateral sclerosis got its name: the clinical-pathologic genius of Jean-Martin Charcot. Archives of Neurology. 2001;58(3):512-515. — JAMA Neurology (Archives of Neurology) full article
- Tofersen (Qalsody) — FDA approval of the first therapy targeting a genetic (SOD1) cause of ALS, April 25, 2023. — U.S. FDA announcement
- Aran F-A and the recognition of progressive muscular atrophy (1850); pre-Charcot history of motor-neuron disease. — PubMed: Aran, progressive muscular atrophy, ALS history
- History and historical perspective of amyotrophic lateral sclerosis. — PubMed: ALS history and Charcot
- Riluzole and the history of disease-modifying ALS therapy. — PubMed: riluzole and ALS survival
- Edaravone (Radicava) in amyotrophic lateral sclerosis. — PubMed: edaravone and ALS
- SOD1 in ALS — gain-of-function mechanism and antisense (tofersen) therapy. — PubMed: SOD1, tofersen, antisense therapy
- El Escorial criteria for the diagnosis of amyotrophic lateral sclerosis. — PubMed: El Escorial diagnostic criteria
- Guam ALS–parkinsonism–dementia complex and environmental hypotheses (BMAA, cycad). — PubMed: Guam ALS-PDC and BMAA
External Authoritative Resources
- NINDS (NIH) — Amyotrophic Lateral Sclerosis (ALS)
- The ALS Association — History of ALS and Lou Gehrig
- MedlinePlus — Amyotrophic Lateral Sclerosis
Connections
- Neurology
- Amyotrophic Lateral Sclerosis (ALS)
- All Conditions
- Parkinson's Disease
- Huntington's Disease
- Multiple Sclerosis
- Myasthenia Gravis
- Alzheimer's Disease