Budd-Chiari Syndrome
- Overview and Definition
- Pathophysiology
- Causes and Risk Factors
- Clinical Presentation
- Diagnosis and Imaging
- Treatment Approach
- Prognosis and Outcomes
- Key Research Papers
- Connections
Overview and Definition
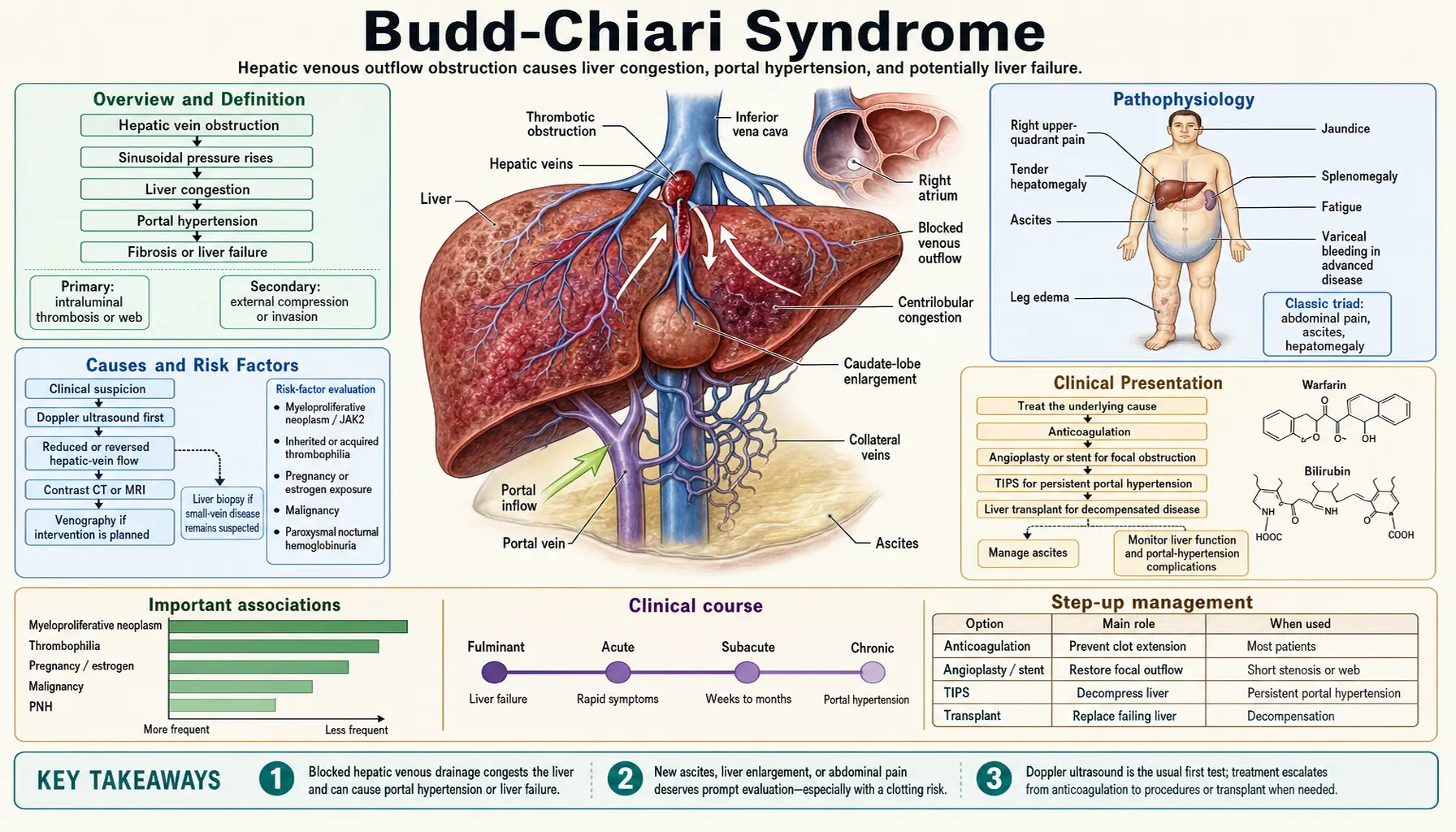
Budd-Chiari Syndrome (BCS) is a rare but serious disorder defined by obstruction of hepatic venous outflow — affecting the hepatic veins, the suprahepatic inferior vena cava (IVC), or both — at any level from the small hepatic venules to the right atrium. The obstruction can be thrombotic, membranous, or caused by extrinsic compression, and it leads uniformly to hepatic venous congestion, sinusoidal hypertension, and ultimately ischemic hepatocellular injury.
First described by the British physician George Budd in 1845 and elaborated by the Austrian pathologist Hans Chiari in 1899, BCS is now understood to be a heterogeneous syndrome rather than a single disease. Its annual incidence is approximately 1 per million in Western populations, though higher rates are reported in Asia and Africa — regions where membranous obstruction of the IVC (MOVC) is more prevalent.
BCS affects adults predominantly, with a peak incidence in the third and fourth decades of life, and women are slightly more affected than men, partly reflecting the contribution of oral contraceptive use and pregnancy to thrombotic risk. Without treatment, acute or subacute BCS carries a poor prognosis, but modern stepwise management — anticoagulation, interventional radiology, and liver transplantation — has dramatically improved survival to more than 70% at 5 years in experienced centers.
Pathophysiology
The central event in BCS is obstruction of hepatic venous drainage. Because the liver receives approximately 25% of cardiac output and has no alternative venous outflow, occlusion of the hepatic veins produces an immediate and severe rise in sinusoidal pressure.
Sinusoidal Hypertension and Congestion
Elevated sinusoidal pressure causes plasma to leak into the space of Disse and then into the peritoneal cavity, producing the characteristic ascites of BCS. Unlike portal hypertension from cirrhosis — where portal pressure rises due to fibrosis and nodular regeneration — the primary abnormality in BCS is venous outflow obstruction. The result is a high-protein, high-gradient ascitic fluid that can accumulate rapidly and is often disproportionately large relative to the degree of liver dysfunction.
Ischemic Hepatocellular Damage
As venous congestion worsens, hepatic arterial inflow becomes relatively inadequate to maintain hepatocyte oxygenation. Centrilobular (zone 3) hepatocytes — furthest from the portal tracts and most dependent on sinusoidal blood flow — undergo ischemic necrosis first. In acute BCS this manifests as centrilobular necrosis and sinusoidal dilatation on biopsy. In chronic BCS, the necrosis evolves into centrilobular fibrosis, nodular regenerative hyperplasia, and eventually cirrhosis.
Portal Hypertension
Hepatic venous congestion and parenchymal fibrosis both raise portal pressure, leading to the full syndrome of portal hypertension: varices (esophageal, gastric, ectopic), splenomegaly, thrombocytopenia, and hepatic encephalopathy in advanced disease. The portal hypertension of BCS is distinct from the "presinusoidal" type seen in portal vein thrombosis — it originates at the sinusoidal level and is compounded by outflow obstruction.
Caudate Lobe Hypertrophy: The Pathognomonic Sign
The caudate lobe is unique among hepatic segments: its small accessory hepatic veins drain directly into the IVC rather than through the three main hepatic veins. When the hepatic veins are obstructed, the caudate lobe alone retains normal venous drainage and therefore escapes the congestion that damages the rest of the liver. The preserved blood supply, combined with compensatory regenerative drive, causes the caudate lobe to undergo dramatic hypertrophy. On imaging, a disproportionately enlarged caudate lobe is virtually pathognomonic of BCS and helps distinguish it from other causes of hepatomegaly and portal hypertension.
Causes and Risk Factors
BCS is classified into primary (intrinsic venous occlusion, almost always thrombotic) and secondary (extrinsic compression or invasion) forms. Identifying the underlying cause is critical both for treatment and for prognosis, since most primary BCS is driven by an identifiable, treatable thrombophilia.
Primary BCS: Thrombotic Occlusion
Approximately 75% of patients with primary BCS have an identifiable prothrombotic disorder. In many patients, multiple concurrent risk factors are present — a finding that argues for comprehensive thrombophilia testing in every case.
Myeloproliferative Neoplasms (MPN) and JAK2 V617F
Myeloproliferative neoplasms are the single most common cause of BCS in Western populations, accounting for 35–50% of cases. Among them, polycythemia vera is most frequent, followed by essential thrombocythemia and primary myelofibrosis. The JAK2 V617F point mutation — a somatic gain-of-function mutation in the Janus kinase 2 gene that drives MPN — is present in 40–50% of patients with "idiopathic" BCS, even when peripheral blood counts appear normal and the full clinical picture of MPN is not yet apparent. Testing for JAK2 V617F is therefore mandatory in every BCS patient. The mutation produces a mildly hypercoagulable, hyperviscous state that preferentially thromboses the hepatic and portal veins.
Antiphospholipid Syndrome (APS)
APS — defined by persistent antiphospholipid antibodies (lupus anticoagulant, anticardiolipin, anti-beta-2-glycoprotein-I) plus arterial or venous thrombosis — accounts for approximately 10–25% of BCS cases. In patients with underlying SLE, the risk is compounded by the lupus-associated hypercoagulable state.
Paroxysmal Nocturnal Hemoglobinuria (PNH)
PNH is a clonal hematopoietic disorder caused by a somatic PIG-A mutation that renders blood cells susceptible to complement-mediated destruction. The hemolysis releases free hemoglobin and promotes nitric oxide scavenging, activating platelets and endothelium. PNH is a particularly important cause of BCS because it is treatable with eculizumab (complement C5 inhibitor), and its diagnosis is often overlooked. Flow cytometry for GPI-anchored proteins (CD55/CD59) should be performed in all BCS patients.
Inherited and Acquired Thrombophilias
- Factor V Leiden (FVL): the most common inherited thrombophilia in Western populations; heterozygous FVL confers a 5–10-fold increased risk of venous thromboembolism; found in 20–30% of BCS patients in some series.
- Prothrombin G20210A mutation: second most common inherited thrombophilia; found in 5–10% of BCS patients.
- Protein C and protein S deficiency: both natural anticoagulants whose deficiency markedly increases thrombotic risk; confounded by liver disease itself (hepatic production), so testing should ideally precede liver injury or be interpreted with caution.
- Antithrombin deficiency: less common but associated with particularly severe thrombotic disease.
- MTHFR mutation and hyperhomocysteinemia: modest independent risk factor.
Oral Contraceptive Pills (OCP) and Pregnancy
Exogenous estrogens — particularly combined oral contraceptives — are an independent risk factor for venous thromboembolism, and hepatic vein thrombosis in women of reproductive age is often the presenting event that reveals an underlying thrombophilia that had previously gone undetected. Pregnancy induces a physiological hypercoagulable state; BCS presenting in the third trimester or postpartum period can be difficult to distinguish from HELLP syndrome or acute fatty liver of pregnancy.
Secondary BCS: Extrinsic Obstruction
Secondary BCS results from structures outside the veins compressing or invading the hepatic venous outflow. Causes include:
- Hepatocellular carcinoma (HCC): HCC invading or compressing the hepatic veins or IVC is a major cause of secondary BCS, particularly in populations with chronic hepatitis B.
- Echinococcal (hydatid) cysts: hepatic hydatid disease can compress major hepatic veins.
- Renal cell carcinoma: can extend via IVC into the suprahepatic IVC.
- Adrenal tumors and retroperitoneal masses: less common causes of extrinsic IVC compression.
Sinusoidal Obstruction Syndrome (SOS) / Veno-Occlusive Disease (VOD)
SOS/VOD is a distinct form of hepatic venous outflow obstruction affecting the terminal hepatic venules and hepatic sinusoids rather than the major hepatic veins. It is most commonly seen after high-dose conditioning chemotherapy for hematopoietic stem cell transplantation (especially busulfan + cyclophosphamide regimens) and after ingestion of pyrrolizidine alkaloids (herbal teas containing comfrey or senecio). SOS does not appear on Doppler ultrasound of the hepatic veins but causes identical sinusoidal congestion and is treated with defibrotide in severe cases.
Clinical Presentation
The clinical presentation of BCS spans a wide spectrum determined by the speed of onset, the completeness of venous occlusion, and the adequacy of collateral venous drainage. Three main clinical forms are recognized.
Acute BCS
Acute BCS develops over days to a few weeks and presents dramatically with the triad of:
- Right upper quadrant (RUQ) pain: often severe and constant, reflecting rapid hepatic capsular distension from edema and congestion.
- Tender hepatomegaly: the liver is enlarged, smooth, and exquisitely tender — in contrast to the hard, irregular liver of cirrhosis.
- Rapidly accumulating ascites: high-protein, serum-ascites albumin gradient (SAAG) >1.1 g/dL ascites develops within days.
Jaundice, nausea, and vomiting are common. A minority of acute BCS cases progress to acute liver failure (ALF) with encephalopathy, coagulopathy, and multiorgan failure — a fulminant presentation that carries very high short-term mortality without emergency liver transplantation.
Subacute and Chronic BCS (Most Common)
Most patients with BCS — approximately 60–70% — present in the subacute or chronic form, characterized by a more insidious course over weeks to months. The cardinal features are:
- Progressive, often massive ascites: the dominant symptom, frequently disproportionate to the degree of liver dysfunction.
- Hepatomegaly: often with the caudate lobe disproportionately enlarged.
- Splenomegaly: reflecting the development of portal hypertension.
- Mild to moderate jaundice: present in some but not all patients; may be absent early.
- Dilated abdominal wall veins: when the IVC is involved, characteristic dilated superficial abdominal veins with upward (cephalad) flow direction distinguish BCS from portal hypertension (where flow is caudal below and cephalad above the umbilicus in caput medusae).
Over time, untreated chronic BCS leads to cirrhosis, portal hypertensive bleeding from esophageal varices, and hepatic encephalopathy.
Fulminant BCS
Fulminant BCS is rare (<5% of cases) and represents acute liver failure occurring within 8 weeks of symptom onset in a patient with no pre-existing liver disease. It carries the worst short-term prognosis and should be listed for emergency liver transplantation immediately, as recovery with medical therapy alone is exceptional.
Incidental (Asymptomatic) BCS
An increasing proportion of BCS is diagnosed incidentally during imaging performed for unrelated reasons, particularly as Doppler ultrasound and cross-sectional abdominal imaging have become routine. Asymptomatic BCS may reflect partial occlusion with adequate collateral drainage, and management must be individualized.
Diagnosis and Imaging
The diagnosis of BCS requires demonstrating obstruction of hepatic venous outflow by imaging. A systematic workup then identifies the underlying cause and guides treatment selection.
Doppler Ultrasound: First-Line Imaging
Doppler ultrasound of the hepatic veins and IVC is the initial investigation of choice. It is rapid, non-invasive, and highly sensitive in experienced hands. Key findings include:
- Absent or reversed hepatic vein flow: the cardinal finding; the normal triphasic waveform of the hepatic veins disappears, replaced by absent, monophasic, or reversed flow.
- Intrahepatic collateral vessels: spider-web or comma-shaped intrahepatic collateral veins visible on color Doppler.
- Caudate lobe hypertrophy: the caudate lobe appears disproportionately enlarged relative to the rest of the liver.
- Hepatic vein thrombosis: the thrombus itself may be directly visualized as echogenic material within the vein lumen.
- IVC narrowing or obstruction: in cases with suprahepatic IVC involvement, the IVC may be compressed by the enlarged caudate lobe or show intrinsic thrombus.
MRI with Contrast
Gadolinium-enhanced MRI provides the most detailed characterization of BCS. It precisely defines the anatomy of hepatic vein and IVC obstruction, identifies parenchymal perfusion defects (heterogeneous enhancement with peripheral patchy hypoperfusion), quantifies caudate lobe enlargement, and detects nodular regenerative hyperplasia or early HCC. MRI is preferred over CT when planning interventional procedures because of its superior soft-tissue contrast without ionizing radiation.
CT with Contrast
Contrast-enhanced CT is often the first cross-sectional study performed and reliably demonstrates hepatic vein occlusion, caudate hypertrophy, and the characteristic "flip-flop" enhancement pattern — peripheral low attenuation (congestion) in the early phase with central enhancement on delayed imaging. CT is also superior to MRI for evaluating the IVC anatomy and planning endovascular intervention.
Thrombophilia Workup
Every patient with confirmed BCS must undergo a comprehensive thrombophilia screen. Testing should be performed before anticoagulation if possible (or after a 4–6 week washout), and results must be interpreted in the context of liver disease, which depletes natural anticoagulants (protein C, S, antithrombin) independently of inherited deficiency. The minimum workup includes:
- JAK2 V617F mutation (PCR or allele-specific PCR) — the single most important test
- PNH screen (flow cytometry for CD55/CD59 on RBCs and granulocytes)
- Antiphospholipid antibodies (lupus anticoagulant, anticardiolipin IgG/IgM, anti-beta-2-GP1 IgG/IgM) — two measurements 12 weeks apart for diagnosis
- Factor V Leiden (PCR)
- Prothrombin G20210A mutation (PCR)
- Protein C activity and antigen
- Protein S total and free antigen (confounded by liver disease and OCP use)
- Antithrombin activity
- MTHFR mutation + fasting homocysteine
- CBC + peripheral smear (polycythemia, thrombocytosis suggesting MPN)
- Serum EPO level (suppressed in polycythemia vera)
- Bone marrow biopsy if MPN suspected clinically or by JAK2 positivity
Liver Biopsy
Liver biopsy is not required for the diagnosis of BCS but provides crucial prognostic information: the degree of centrilobular necrosis, fibrosis, and nodular regeneration helps determine urgency of intervention and guides transplant listing decisions. Transjugular liver biopsy (performed at the same session as hepatic venography) is preferred over percutaneous biopsy to avoid bleeding complications in patients with coagulopathy and ascites.
Treatment Approach
Treatment of BCS follows a stepwise algorithm that aims to relieve venous outflow obstruction, prevent thrombus extension, treat the underlying cause, and manage complications of portal hypertension. The EASL Clinical Practice Guidelines (2016) and AASLD recommendations provide the current framework.
Step 1: Anticoagulation (All Patients)
Anticoagulation is the cornerstone of BCS management and should be started immediately in all patients with confirmed thrombotic BCS, including those with thrombocytopenia or mild coagulopathy. There is no evidence that thrombocytopenia (even platelet counts >50 × 10⁹/L) increases bleeding risk sufficiently to withhold anticoagulation in BCS.
- Initiation: Low molecular weight heparin (LMWH) is preferred for bridging; start on the day of diagnosis.
- Maintenance: Vitamin K antagonists (warfarin, target INR 2.0–3.0) have been the traditional long-term choice. Direct oral anticoagulants (DOACs) — rivaroxaban, apixaban — are increasingly used but evidence specific to BCS is limited; they are preferred in patients with concurrent MPN or APS (for whom warfarin dosing is complex).
- Duration: Lifelong anticoagulation is recommended for almost all BCS patients because the underlying thrombophilia is rarely reversible, and the risk of recurrent thrombosis is high.
Step 2: Treat the Underlying Cause
Simultaneous treatment of the underlying prothrombotic condition is essential:
- MPN (polycythemia vera/ET/PMF): cytoreduction with hydroxyurea (phlebotomy for PV to maintain hematocrit <45%); JAK inhibitors (ruxolitinib) for refractory cases; consider allogeneic stem cell transplantation for high-risk MF.
- PNH: eculizumab (anti-C5 monoclonal antibody) dramatically reduces thrombotic risk and improves outcomes in PNH-associated BCS; it does not replace anticoagulation but is synergistic.
- APS: warfarin anticoagulation (target INR 2.5–3.5 for high-risk APS); hydroxychloroquine as adjunctive agent.
Step 3: Venous Recanalization
When anticoagulation alone does not achieve adequate clinical improvement — defined as failure to reduce ascites, improve liver function tests, or prevent hepatic decompensation over 3–6 months — venous recanalization by interventional radiology is the next step.
- Percutaneous transluminal angioplasty (PTA) ± stenting: for short-segment or membranous hepatic vein or IVC obstruction, balloon dilation with or without metallic stent placement can achieve dramatic and durable relief. Success rates of 70–90% with low procedural mortality make this the preferred intervention when anatomy is favorable.
- Catheter-directed thrombolysis: used adjunctively in acute thrombosis where thrombus is fresh and accessible.
Step 4: TIPS (Transjugular Intrahepatic Portosystemic Shunt)
TIPS is the pivotal intervention for BCS patients who fail anticoagulation alone and are not candidates for recanalization, or who have significant portal hypertension complications (refractory ascites, variceal bleeding). By creating a low-resistance channel between the portal vein and the IVC, TIPS bypasses the obstructed hepatic veins and decompresses both the portal and sinusoidal circulations simultaneously.
- Technical success: achievable in 95% of BCS cases with experienced operators, though the procedure is technically more challenging than TIPS for cirrhosis due to hepatic vein occlusion.
- Clinical outcomes: 5-year transplant-free survival exceeds 70% in BCS patients treated with TIPS, superior to historical controls managed with surgical shunting.
- TIPS dysfunction: covered stent-grafts (PTFE-covered) have dramatically reduced TIPS dysfunction rates compared to bare-metal stents; annual Doppler surveillance is standard.
Step 5: Liver Transplantation
Liver transplantation is reserved for patients with fulminant BCS (acute liver failure without prior liver disease), those who fail TIPS (TIPS occlusion unresponsive to revision), or those with end-stage cirrhosis secondary to chronic BCS. Post-transplant outcomes in BCS are excellent — 5-year survival exceeds 70% — better than for most other indications for transplantation, reflecting the relative youth of BCS patients and the absence of oncologic or metabolic comorbidities in many cases. Lifelong anticoagulation post-transplant is essential because the underlying thrombophilia persists.
Management of Complications
- Ascites: salt restriction (88 mmol/day), aldosterone antagonists (spironolactone 100–400 mg/day), loop diuretics; large-volume paracentesis with albumin replacement for diuretic-refractory ascites.
- Variceal hemorrhage: non-selective beta-blockers (propranolol/carvedilol) for primary prophylaxis; endoscopic band ligation for bleeding varices; TIPS for refractory bleeding.
- HCC surveillance: BCS with cirrhosis confers HCC risk; 6-monthly ultrasound ± AFP is standard.
Prognosis and Outcomes
The prognosis of BCS has been transformed over the past two decades by the adoption of the stepwise treatment algorithm, widespread use of TIPS, and better recognition of the underlying MPN/JAK2 etiology. Historical untreated BCS carried 3-year mortality exceeding 85%; modern series report 5-year survival of 55–87% depending on disease severity, cause, and access to interventional and transplant services.
Prognostic Scores
The Rotterdam BCS Prognostic Score uses four variables — age, prothrombin time, serum bilirubin, and ascites — to stratify patients into low, intermediate, and high risk. The BCS-TIPS Prognostic Score, developed by the European Network for Vascular Disorders of the Liver (EN-Vie), incorporates bilirubin, creatinine, and INR to predict 1-year mortality after TIPS and identifies patients best served by early transplant listing.
Predictors of Poor Outcome
- Fulminant presentation with acute liver failure
- High Model for End-Stage Liver Disease (MELD) score at presentation
- Complete occlusion of all three major hepatic veins plus IVC
- Absence of identifiable and treatable underlying thrombophilia
- Concurrent portal vein thrombosis (extends the occlusion upstream)
- Development of HCC in the setting of BCS-related cirrhosis
Long-Term Considerations
Survivors of BCS require lifelong anticoagulation and monitoring for: (1) TIPS dysfunction (annual Doppler); (2) MPN progression (annual CBC, bone marrow biopsy if counts evolve); (3) PNH clone expansion; (4) HCC if cirrhosis is present; and (5) recurrent thrombosis in other sites. Women with BCS who wish to conceive require multidisciplinary planning — therapeutic anticoagulation throughout pregnancy and close obstetric monitoring are mandatory, but successful pregnancies are reported.
Key Research Papers
- Janssen HL et al. Hepatology 2004 — Comprehensive review of Budd-Chiari syndrome: clinical features, etiology, and management strategies in Western patients — Search PubMed
- Valla DC. J Hepatol 2009 — Hepatic vein thrombosis: pathogenesis, clinical presentation, and diagnosis in the modern era — Search PubMed
- Hernandez-Gea V et al. Gastroenterology 2019 — Current management strategies for Budd-Chiari syndrome: stepwise algorithm from anticoagulation through liver transplantation — Search PubMed
- Darwish Murad S et al. Gastroenterology 2009 — Etiology, management, and outcome of the Budd-Chiari syndrome in a large multinational European cohort (EN-Vie study) — Search PubMed
- Smalberg JH et al. Blood 2012 — Myeloproliferative neoplasms and the JAK2 V617F mutation in patients with Budd-Chiari syndrome and portal vein thrombosis — Search PubMed
- Primignani M. Dig Liver Dis 2010 — Portal vein thrombosis, relevance on liver cirrhosis, and MPN: clinical overlap and shared mechanisms — Search PubMed
- Qi X et al. J Gastroenterol Hepatol 2013 — TIPS for Budd-Chiari syndrome: systematic review of patency, clinical response, and survival outcomes — Search PubMed
- Trebicka J et al. J Hepatol 2014 — TIPS for Budd-Chiari syndrome: long-term outcomes and predictors of failure and transplant-free survival — Search PubMed
- Seijo S et al. Gastroenterology 2013 — Role of the caudate lobe in Budd-Chiari syndrome: compensatory hypertrophy, vascular anatomy, and clinical significance — Search PubMed
- Northup PG et al. Gastroenterology 2014 — Anticoagulation in patients with cirrhosis and hepatic vein thrombosis: evidence base, safety, and outcomes — Search PubMed
- Zeitoun G et al. J Hepatol 1999 — Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting — Search PubMed
- De Gottardi A et al. J Hepatol 2017 — EASL Clinical Practice Guidelines on vascular diseases of the liver, including Budd-Chiari syndrome: diagnosis, workup, and stepwise management — Search PubMed
Connections
- Nephrology & Hepatology
- Cirrhosis
- Primary Biliary Cholangitis
- Autoimmune Hepatitis
- Hepatic Encephalopathy
- Liver Disease
- Deep Vein Thrombosis
- Disseminated Intravascular Coagulation
- Essential Thrombocythemia
- Coagulation Panel