Loeys-Dietz Syndrome
- Overview and Genetics
- LDS Subtypes 1–5
- Vascular Manifestations
- Craniofacial and Skeletal Features
- Skin and Joint Findings
- Diagnosis and Genetic Testing
- Surveillance and Imaging
- Medical and Surgical Treatment
- Prognosis and Natural History
- Key Research Papers
- Connections
- Featured Videos
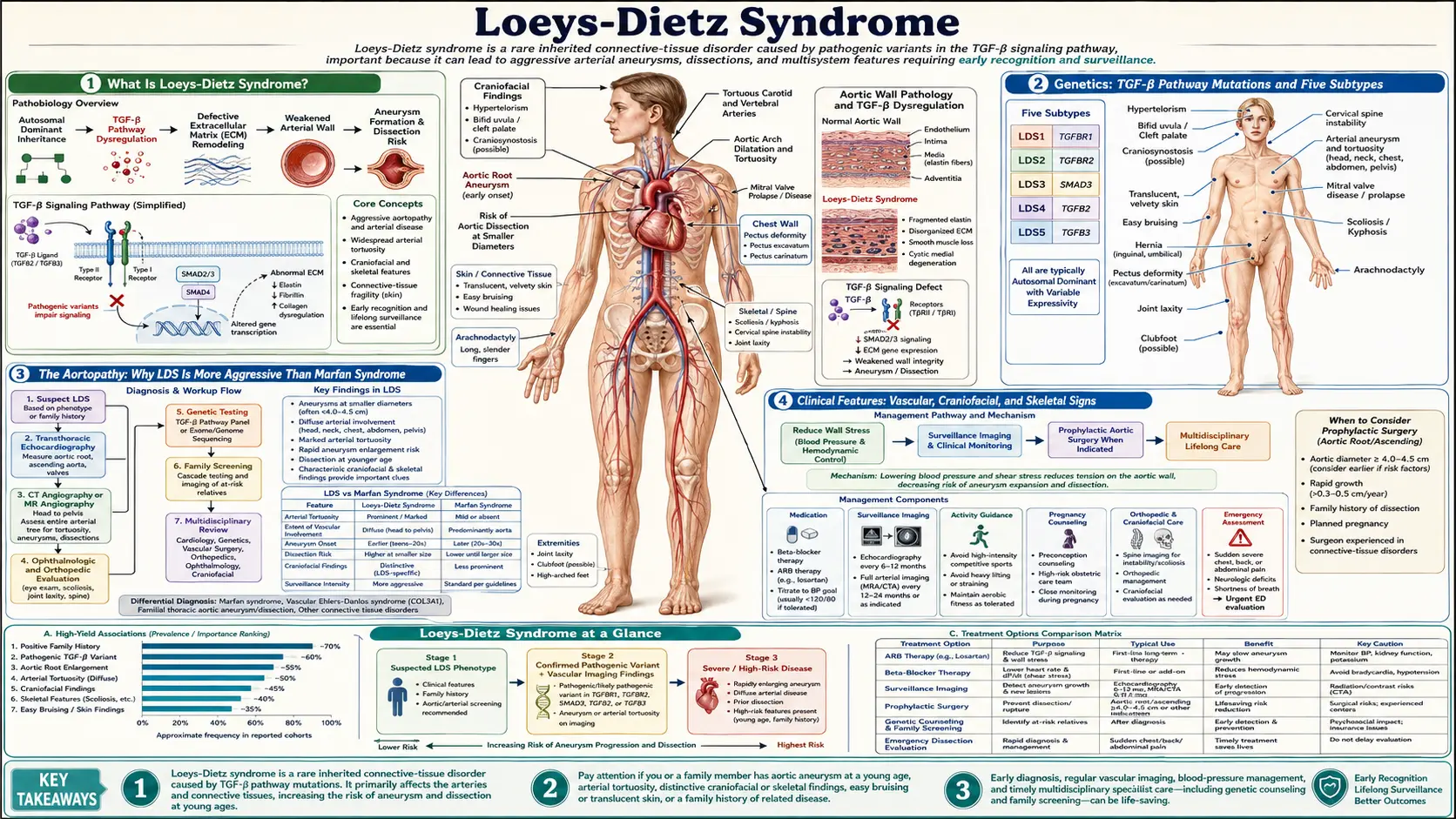
Overview and Genetics
Loeys-Dietz syndrome (LDS) is an autosomal dominant connective tissue disorder caused by mutations in the TGF-β signaling pathway. First described in 2005 by Bart Loeys and Harry Dietz, LDS is characterized by a triad of arterial aneurysms and tortuosity, skeletal manifestations, and craniofacial anomalies. Despite its relatively recent recognition, LDS is now understood to carry a more aggressive vascular phenotype than Marfan syndrome, with arterial dissection occurring at smaller aortic diameters and affecting branch arteries throughout the body.
The hallmark molecular defect involves mutations in genes encoding TGF-β receptors or downstream signaling molecules. Paradoxically, despite loss-of-function mutations in these receptors, tissue analysis shows paradoxical TGF-β hyperactivation — excessive phosphorylation of SMAD2/3 and elevated connective tissue growth factor (CTGF). This counterintuitive biology, known as the "TGF-β paradox," explains why TGF-β pathway inhibition with losartan has therapeutic rationale even though the primary mutations appear to reduce receptor function. The current model suggests that heterozygous receptor haploinsufficiency triggers compensatory upregulation of alternative TGF-β signaling routes, ultimately resulting in net pathway overdrive in vascular wall cells.
LDS affects approximately 1 in 50,000–100,000 individuals, though prevalence is likely underestimated because it was previously misclassified as Marfan syndrome, vascular Ehlers-Danlos syndrome, or Shprintzen-Goldberg syndrome. Approximately 75% of cases arise from de novo mutations; 25% are familial. Penetrance is nearly complete, though expressivity varies significantly even within families.
LDS Subtypes 1–5
The 2014 revised classification divides LDS into five subtypes based on the causative gene:
- LDS Type 1 — TGFBR2 (chromosome 3p24.1): The originally described subtype. Mutations in the TGF-β receptor type II gene. Associated with the full classic triad including hypertelorism, bifid uvula or cleft palate, and aggressive aortic disease. Typically the most severe phenotype.
- LDS Type 2 — TGFBR1 (chromosome 9q22.33): Mutations in TGF-β receptor type I. Phenotypically overlapping with Type 1 but sometimes milder craniofacial involvement. Skin and joint laxity can be pronounced; some cases were previously reported as "Furlong syndrome" or familial thoracic aortic aneurysm.
- LDS Type 3 — SMAD3 (chromosome 15q22.33): Also called aneurysms-osteoarthritis syndrome (AOS). Mutations in the intracellular SMAD3 mediator. Notable for early-onset osteoarthritis in addition to aneurysms, with less prominent craniofacial features. Joint disease can be debilitating independent of aortic complications.
- LDS Type 4 — TGFB2 (chromosome 1q41): Mutations in the TGF-β2 ligand itself. Milder craniofacial features; skeletal and vascular manifestations present but may be delayed in onset. Some overlap with Marfan syndrome features including lens dislocation reported in rare cases.
- LDS Type 5 — TGFB3 (chromosome 14q24.3): Mutations in the TGF-β3 ligand. The rarest subtype; described in families with mitral valve disease, aortic aneurysm, and skeletal findings. Craniofacial features less prominent than LDS Types 1–2.
All five subtypes share the core risk of life-threatening aortic and branch arterial disease, mandating aggressive surveillance and low surgical thresholds regardless of subtype.
Vascular Manifestations
The vascular phenotype is the most dangerous aspect of LDS and distinguishes it from other heritable connective tissue disorders. Key features include:
Aortic root aneurysm: The aortic root (sinus of Valsalva) is the most common site of aneurysm, seen in the majority of LDS patients. Unlike Marfan syndrome, where aneurysms rarely dissect below a diameter of 50mm, LDS patients may dissect at 40mm or even smaller. Prophylactic aortic root replacement is recommended at 42–45mm in most centers (compared to 50mm for Marfan), and some guidelines recommend surgery at 40mm for individuals with rapidly enlarging aneurysms, family history of dissection, or pregnancy planning.
Arterial tortuosity (pathognomonic): Widespread tortuosity of medium and large arteries — including the carotid, vertebral, subclavian, renal, splenic, and iliac vessels — is a hallmark finding not typically seen in Marfan syndrome or vEDS. On CT angiography, tortuous arteries often appear as "corkscrew" or "S-shaped" vessels. This tortuosity reflects abnormal extracellular matrix in arterial walls and is an independent predictor of vascular risk. CT or MR angiography must cover the entire aorta and branch arteries, not just the chest.
Branch artery aneurysms: Unlike Marfan syndrome (which primarily affects the aortic root), LDS causes aneurysms and dissections in branch arteries including the celiac, mesenteric, renal, subclavian, and intracranial vessels. Spontaneous cervical artery dissection (carotid and vertebral) causing stroke has been reported in young LDS patients without preceding trauma.
Dissection at smaller diameters: A defining and clinically critical difference from Marfan syndrome. LDS patients tolerate far less aortic dilation before dissection risk becomes prohibitive. Surgical planning must account for this compressed risk window.
Intracranial aneurysms: Reported in a subset of LDS patients, warranting brain MRA in selected individuals, particularly those with severe arterial tortuosity elsewhere.
Craniofacial and Skeletal Features
LDS Types 1 and 2 in particular display a distinctive craniofacial gestalt that aids clinical recognition:
- Hypertelorism: Wide-spaced eyes (increased interpupillary distance) is one of the most consistent craniofacial findings. Orbital dystopia may be present. The degree of hypertelorism correlates roughly with phenotype severity in some studies.
- Bifid uvula or cleft palate: Midline palatal defects range from a bifid (split) uvula — visible on oral examination — to frank cleft palate requiring surgical repair in infancy. Present in approximately 50–60% of LDS Types 1–2.
- Craniosynostosis: Premature fusion of cranial sutures, seen in a subset of patients. Turricephaly (tower skull), scaphocephaly, or plagiocephaly may result. Craniosynostosis in the context of aortic aneurysm should prompt LDS testing.
- Retrognathia / micrognathia: Small or recessed jaw, contributing to dental crowding and sometimes airway issues requiring orthodontic or surgical management.
Skeletal manifestations are common across all subtypes and include:
- Pectus excavatum or pectus carinatum (chest wall deformity)
- Scoliosis, which can be progressive and may require bracing or surgery
- Arachnodactyly (long, slender fingers) and dolichostenomelia (long limbs) — overlapping with Marfan syndrome
- Joint laxity and instability, particularly at the wrist, fingers, and ankles
- Cervical spine instability (atlantoaxial or subaxial), a potentially serious complication requiring evaluation before general anesthesia
- Club foot (talipes equinovarus) in neonates and infants
- Dural ectasia (widening of the spinal dural sac), detected on lumbar MRI
In LDS Type 3 (SMAD3), early-onset osteoarthritis affecting the knees, hips, and spine may be the presenting complaint, often leading to incorrect diagnoses of inflammatory arthritis before vascular findings prompt genetic testing.
Skin and Joint Findings
While skin fragility is not as severe as in vascular Ehlers-Danlos syndrome (COL3A1), LDS patients frequently have skin and connective tissue findings that reflect the underlying matrix defect:
- Translucent skin: Thin skin through which underlying veins are easily visible, particularly on the chest, forearms, and hands. This reflects reduced collagen integrity in the dermis.
- Easy bruising: Subcutaneous hemorrhage with minor trauma is common. Unlike vEDS, bruising in LDS rarely predicts arterial rupture but can be alarming to patients and parents.
- Stretch marks (striae): Striae distensae in atypical locations (not related to obesity or rapid growth) — over the chest, flanks, and lower back — seen in a subset of patients.
- Delayed wound healing: Incision healing may be prolonged; surgeons managing LDS patients should be aware of this risk, particularly after aortic surgery.
- Generalized joint hypermobility: Elevated Beighton score is common, though not universal. Joint instability at the shoulders, hips, knees, and ankles can limit function and contribute to chronic musculoskeletal pain.
- Hernias: Inguinal, umbilical, and incisional hernias occur at elevated rates due to reduced connective tissue tensile strength.
In LDS Type 3 (SMAD3 mutations), the skin and joint phenotype may be particularly prominent, with some patients meeting criteria for a hypermobile-type connective tissue disorder in addition to the osteoarthritis that defines this subtype.
Diagnosis and Genetic Testing
LDS diagnosis rests on the combination of clinical recognition and molecular confirmation:
Clinical recognition triggers: Any of the following should prompt LDS evaluation — aortic root aneurysm in a young person, bifid uvula with aortic disease, arterial tortuosity on imaging, family history of aortic dissection or sudden death, or a connective tissue disorder phenotype that doesn't clearly fit Marfan syndrome or EDS.
Molecular genetic panel testing: A multi-gene panel covering TGFBR1, TGFBR2, SMAD3, TGFB2, and TGFB3 is now standard, typically alongside genes for Marfan syndrome (FBN1), vEDS (COL3A1), MASS phenotype, and familial thoracic aortic aneurysm syndromes (ACTA2, MYH11, MYLK). Turnaround is 3–4 weeks for commercial panels.
Variants of uncertain significance (VUS): A common challenge in LDS testing. Segregation studies in affected family members, protein functional assays, and population database frequency (gnomAD) all contribute to reclassification over time. Functional RNA studies may be needed for splicing variants.
Echocardiography as first imaging step: Transthoracic echocardiography should be performed at diagnosis to assess aortic root diameter. CT angiography or MR angiography of the full aorta and branch arteries is essential for baseline mapping, given the predilection for aneurysms beyond the aortic root.
First-degree family screening: All first-degree relatives should be offered genetic testing once a pathogenic variant is identified. Imaging should begin in confirmed carriers regardless of symptoms, as LDS can be asymptomatic until a catastrophic dissection.
Differential diagnosis: LDS must be distinguished from Marfan syndrome (FBN1), vascular EDS (COL3A1), Shprintzen-Goldberg syndrome (SKI), and MASS phenotype. Key distinguishing features of LDS include arterial tortuosity, bifid uvula, and dissection at smaller diameters.
Surveillance and Imaging
LDS requires lifelong, proactive imaging surveillance — more aggressive than for Marfan syndrome — because aneurysms can progress rapidly and occur at unexpected locations:
- Annual comprehensive aortic imaging: CT angiography or MR angiography (MRA preferred in young patients to minimize radiation) from the cerebrovascular circulation to the pelvis. This full-body approach captures branch artery aneurysms in the celiac, renal, mesenteric, subclavian, and iliac territories that would be missed by echocardiography alone.
- Echocardiography: Annual transthoracic echo to follow aortic root and proximal ascending aorta dimensions with high reproducibility. Echo complements but does not replace cross-sectional aortic imaging.
- Brain MRA: Recommended at baseline to screen for intracranial aneurysms, particularly in patients with severe arterial tortuosity; repeat interval individualized based on findings.
- Cervical spine MRI or CT: At baseline to evaluate for cervical instability (C1-C2 subluxation), especially prior to general anesthesia, contact sports, or invasive procedures.
- Ophthalmologic evaluation: Lens dislocation is uncommon in LDS but reported in LDS Type 4 (TGFB2); baseline slit-lamp examination is reasonable.
- More frequent imaging during pregnancy: Aortic diameter increases during pregnancy due to volume loading. Monthly or bimonthly echocardiography during pregnancy and 6 months postpartum is recommended. Epidural anesthesia is preferred over general anesthesia to reduce hemodynamic stress at delivery.
Patients should be counseled to avoid isometric exercise (heavy weight lifting), contact sports, competitive athletics, and Valsalva maneuvers, which transiently increase aortic wall stress. Low-impact aerobic exercise (swimming, walking, cycling at moderate intensity) is encouraged for cardiovascular health.
Medical and Surgical Treatment
Losartan (angiotensin II receptor blocker): The primary medical therapy for LDS based on TGF-β pathway biology. Losartan blocks the AT1 receptor, reducing TGF-β signaling in the aortic wall. Preclinical studies in Tgfbr1 and Tgfbr2 mutant mice demonstrated dramatic reduction in aortic root growth rates with losartan. Clinical evidence in human LDS is extrapolated from Marfan trials (COMPARE and Pediatric Heart Network trials) and LDS registry data. Current practice is to initiate losartan at diagnosis and dose to maximal tolerated blood pressure reduction (target systolic BP <120 mmHg). Beta-blockers (atenolol) are sometimes added to further reduce aortic wall shear stress.
Prophylactic aortic surgery — lower threshold than Marfan: The recommended threshold for prophylactic aortic root replacement in LDS is 42–45mm, compared to 50mm for Marfan syndrome. This lower cutoff reflects the observation that LDS patients dissect at smaller diameters. The Valve-Sparing Root Replacement (David procedure or Yacoub procedure) is preferred when the aortic valve leaflets are morphologically normal, preserving the native valve and avoiding lifelong anticoagulation. Root replacement with a composite valve-graft (Bentall procedure) is used when the valve is abnormal or in urgent cases.
Branch artery interventions: Endovascular stent-graft repair of branch artery aneurysms (celiac, renal, iliac) is feasible but complicated by arterial tortuosity, which can impede catheter delivery and stent landing. Open surgical repair is often preferred for younger LDS patients when anatomy permits.
Pregnancy management: Women with LDS face substantial risk during pregnancy due to cardiovascular stress and progesterone-mediated connective tissue softening. Preconception counseling should include aortic sizing; many centers recommend prophylactic aortic surgery before planned pregnancy if the root diameter approaches 40mm. Delivery should be planned in a center with cardiac surgery capability.
Pain management and rehabilitation: Chronic musculoskeletal pain is common due to joint hypermobility, scoliosis, and osteoarthritis (particularly in LDS Type 3). Physical therapy emphasizing core stabilization and low-impact strengthening improves joint stability without increasing vascular stress. NSAIDs should be used cautiously given potential effects on platelet function and renal protective prostaglandins in patients on aggressive antihypertensive therapy.
Neurosurgical and orthopedic interventions: Cervical instability may require posterior cervical fusion. Severe scoliosis may require spinal instrumentation. Craniosynostosis in infants requires neurosurgical cranioplasty. Coordination between cardiovascular, orthopedic, and neurosurgical teams is essential.
Prognosis and Natural History
Before widespread recognition of LDS and implementation of aggressive surveillance and early surgery, outcomes were poor. Early registry data showed a median age of death of approximately 26 years for individuals with TGFBR1 or TGFBR2 mutations. With modern management — including early genetic diagnosis, comprehensive imaging surveillance, prophylactic surgery at lower thresholds, and medical therapy — survival has improved substantially, though long-term natural history data for proactively managed cohorts are still accumulating.
Prognosis varies by subtype. LDS Types 1 and 2 (TGFBR2 and TGFBR1) carry the most aggressive vascular phenotype. LDS Type 3 (SMAD3 / aneurysms-osteoarthritis syndrome) tends to have slower aortic progression but carries significant morbidity from osteoarthritis. LDS Types 4 and 5 (TGFB2 and TGFB3) appear intermediate in severity, though fewer patients have been followed long-term.
Key adverse prognostic factors include:
- Rapidly expanding aortic root (>3–5 mm/year)
- Aortic root diameter >45mm without surgical intervention
- Family history of aortic dissection at young age
- TGFBR2 mutations (historically associated with more aggressive vascular disease)
- Extensive branch artery aneurysms at baseline imaging
- Cervical arterial tortuosity and intracranial aneurysms
Psychological burden is significant in LDS. The combination of a life-threatening vascular diagnosis, activity restrictions, anticipatory anxiety about aortic events, and complex surgical histories contributes to high rates of depression and anxiety. Patient support organizations (LDS Alliance) and psychological support should be integrated into multidisciplinary care.
Quality of life research in LDS remains limited compared to Marfan syndrome. Registry-based outcomes studies — particularly from the GenTAC (Genetically Triggered Thoracic Aortic Conditions) consortium and the LDS Registry at Johns Hopkins — are the primary evidence base for natural history data.
Key Research Papers
- Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nature Genetics. 2005;37(3):275–281. PMID: 15731757. The founding description of LDS, defining the syndrome and its molecular basis.
- Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. New England Journal of Medicine. 2006;355(8):788–798 — Search PubMed. Expanded clinical characterization of 52 LDS patients; defined the aggressive natural history and surgical recommendations.
- van de Laar IM, Oldenburg RA, Pals G, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nature Genetics. 2011;43(2):121–126. PMID: 21217753. Discovery paper establishing SMAD3 mutations as the cause of LDS Type 3 / aneurysms-osteoarthritis syndrome.
- Lindsay ME, Schepers D, Bolar NA, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nature Genetics. 2012;44(8):922–927. PMID: 22772371. Defined LDS Type 4 caused by TGFB2 ligand mutations.
- Boileau C, Guo DC, Hanna N, et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nature Genetics. 2012;44(8):916–921. PMID: 22772368. Parallel TGFB2 discovery paper; noted phenotypic overlap with Marfan syndrome.
- Guo DC, Regalado ES, Gong L, et al. LOF mutations in TGFB3 cause familial thoracic aortic aneurysms and dissections. American Journal of Human Genetics. 2015;97(4):611–619 — Search PubMed. Established TGFB3 mutations as the cause of LDS Type 5.
- Rodrigues VJ, Elsayed S, Loeys BL, Dietz HC, Yousem DM. Neuroradiologic manifestations of Loeys-Dietz syndrome type 1. AJNR American Journal of Neuroradiology. 2009;30(8):1614–1619 — Search PubMed. Characterizes intracranial vascular and skull-base findings in LDS, supporting brain MRA screening.
- Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117–121 — Search PubMed. Foundational preclinical study establishing the TGF-β → losartan therapeutic rationale used in LDS.
- Williams JA, Loeys BL, Nwakanma LU, et al. Early surgical experience with Loeys-Dietz: a new syndrome of aggressive thoracic aortic aneurysm disease. Annals of Thoracic Surgery. 2007;83(2):S757–S763 — Search PubMed. Surgical outcomes from the first cohort managed with aggressive prophylactic surgery; established the 40–42mm threshold rationale.
- Schepers D, Tortora G, Morisaki H, et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Human Mutation. 2018;39(5):621–634 — Search PubMed. Comprehensive mutation database and genotype-phenotype correlations across all LDS subtypes.
- Franken R, El Morabit A, de Waard V, et al. Increased aortic tortuosity indicates a more severe aortic phenotype in adults with Marfan syndrome. International Journal of Cardiology. 2015;194:7–12 — Search PubMed. Quantifies arterial tortuosity as an imaging biomarker correlating with disease severity; methodology applicable to LDS.
- Oderich GS, Panneton JM, Bower TC, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. Journal of Vascular Surgery. 2005;42(1):98–106 — Search PubMed. Surgical outcomes in vascular connective tissue disorders; provides comparative context for LDS vascular intervention thresholds and outcomes.
PubMed searches for additional research:
Loeys-Dietz syndrome aorta TGFBR1 TGFBR2 aneurysm Loeys-Dietz losartan treatment SMAD3 aortic aneurysm osteoarthritis
Connections
- Genetics
- Ehlers-Danlos Syndrome
- Marfan Syndrome
- Aortic Aneurysm
- Aortic Dissection
- Stroke
- POTS (Postural Orthostatic Tachycardia Syndrome)
- Scoliosis
- Diseases
- Lab Tests