Pemphigus Vulgaris
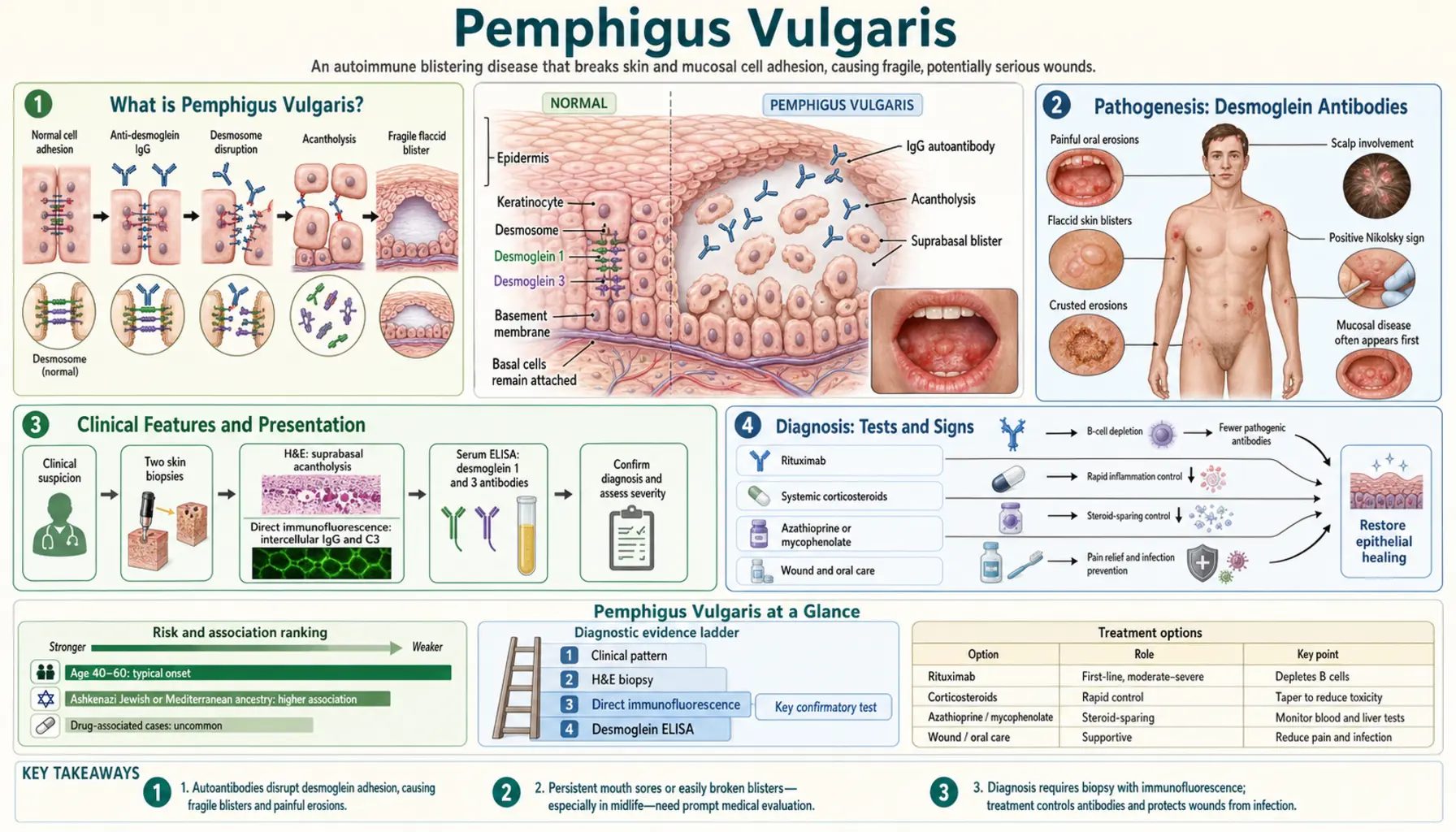
Table of Contents
- What is Pemphigus Vulgaris?
- Pathogenesis: Desmoglein Antibodies
- Clinical Features and Presentation
- Diagnosis: Tests and Signs
- Pemphigus Variants
- Induction Therapy: Corticosteroids
- Rituximab: The Modern Standard
- Maintenance and Monitoring
- Paraneoplastic Pemphigus
- Prognosis and Quality of Life
- Research Papers
- Connections
- Featured Videos
What is Pemphigus Vulgaris?
Pemphigus vulgaris (PV) is a rare, potentially life-threatening autoimmune blistering disease of the skin and mucous membranes. The immune system generates IgG autoantibodies against desmoglein proteins — the molecular "glue" that holds skin cells together — causing them to lose cohesion and separate. The result is fragile, flaccid blisters that rupture easily, leaving raw, painful erosions that heal slowly.
PV has a worldwide incidence of 1–5 per 100,000 per year, with higher rates in Jewish, Indian, and Mediterranean populations where certain HLA haplotypes predispose. It most commonly presents in the 4th–6th decade, affecting men and women roughly equally. Before corticosteroids were available, PV was fatal in approximately 75% of patients within 2–5 years from infections, fluid loss, and sepsis. Today, with modern immunosuppressive therapy — and particularly with the introduction of rituximab — mortality has fallen to under 5%, though the disease remains seriously disabling.
The hallmark clinical clue that distinguishes PV from other blistering disorders: oral involvement in 90% of patients, often months before skin blisters appear. A patient with painful, recalcitrant mouth sores not responding to standard treatments should raise clinical suspicion for PV.
Pathogenesis: Desmoglein Antibodies
Desmogleins (DSG) are transmembrane glycoproteins that form the adhesive core of desmosomes — the intercellular junctions that rivet keratinocytes (skin cells) together. Without functional desmogleins, keratinocytes lose adhesion and separate — a process called acantholysis (loss of cell-to-cell cohesion within the epidermis).
Two desmogleins are targeted in pemphigus:
- Desmoglein 3 (DSG3): Highly expressed in mucous membranes and the lower epidermis. Anti-DSG3 IgG causes mucosal blistering. Present in nearly all PV patients.
- Desmoglein 1 (DSG1): Highly expressed in the upper epidermis and skin. Anti-DSG1 IgG adds cutaneous involvement. Present in about 50–60% of PV patients and in all patients with pemphigus foliaceus.
The desmoglein compensation hypothesis explains the anatomical distribution: in the upper epidermis, DSG1 is abundant and can compensate if DSG3 is targeted (no skin blistering from anti-DSG3 alone). In mucosa, DSG1 is scarce so DSG3 alone provides cohesion — thus anti-DSG3 antibodies always cause mucosal disease. When both anti-DSG3 and anti-DSG1 are present, both mucosa and skin blister.
The mechanism of antibody action is direct: anti-DSG IgG sterically blocks desmoglein-desmoglein interactions and triggers intracellular signaling cascades (involving p38 MAPK and RhoA) that further weaken keratinocyte adhesion. Complement activation plays a secondary role. The separation occurs just above the basal cell layer, creating the "suprabasal" split that is histologically definitive for PV.
Genetic predisposition involves HLA class II alleles (DRB1*0402, DQB1*0503 confer the highest risk). Environmental triggers include certain drugs (captopril, penicillamine, rifampin) and, rarely, viral infections.
Clinical Features and Presentation
Oral Mucosal Involvement (First Sign in 90%)
Oral erosions are the presenting complaint in the majority of PV patients and may precede skin blistering by weeks to months — sometimes more than a year. The erosions are painful, irregular, and slow to heal. They typically affect the buccal mucosa, gingiva, palate, and posterior pharynx. Patients frequently report difficulty eating, speaking, and swallowing. These lesions are commonly misdiagnosed as aphthous ulcers, candidiasis, or herpes before PV is considered.
Skin Blisters
Cutaneous blisters in PV are characteristically flaccid (thin-walled, easily ruptured) — in sharp contrast to the tense blisters of bullous pemphigoid, which arise beneath the epidermis and are much harder to break. PV blisters arise within the epidermis and the overlying roof is so fragile it ruptures spontaneously or with minimal trauma, leaving extensive raw erosions. Healing is slow, and secondary bacterial infection (particularly Staphylococcus aureus) is common and a major cause of morbidity.
Blisters can appear anywhere on the body but favor the scalp, face, chest, back, and intertriginous areas. The trunk is commonly involved.
Other Mucosal Sites
Beyond the oral cavity, PV can involve the conjunctiva (ocular mucosa), nasal mucosa (epistaxis, crusting), larynx (hoarseness, aspiration risk), esophagus (dysphagia), and anogenital mucosa (painful erosions).
Diagnosis: Tests and Signs
Nikolsky Sign
Lateral finger pressure on normal-appearing perilesional skin shears off the superficial epidermis, extending the blister laterally. Positive Nikolsky sign reflects active acantholysis and is highly characteristic of pemphigus (and other acantholytic disorders) — absent in bullous pemphigoid where the epidermis is firmly anchored to dermis. The Asboe-Hansen sign (pressing down on an intact blister causes it to extend laterally) is related.
Tzanck Smear
A quick bedside test: scraping the base of a freshly ruptured blister and staining with Giemsa or Wright stain reveals acantholytic "Tzanck cells" — rounded, detached keratinocytes with large nuclei and abundant cytoplasm. Positive in pemphigus but not specific (also seen in herpes infections). It can provide rapid supportive evidence while awaiting biopsy.
Skin Biopsy with Histology
Biopsy of a freshly formed blister (or blister edge) shows the pathognomonic suprabasal acantholytic split: a cleft just above the basal cell layer with tombstone-row basal cells still adherent to the basement membrane and rounded acantholytic cells floating free in the blister cavity.
Direct Immunofluorescence (DIF) — Gold Standard
Biopsy of perilesional skin stained with fluorescently tagged anti-IgG antibodies shows the characteristic "fishnet" or "chicken wire" intercellular IgG deposition throughout the epidermis — IgG outlining the surfaces of every keratinocyte in a network pattern. This pattern is pathognomonic for pemphigus and is the definitive diagnostic test. C3 complement may co-deposit.
Indirect Immunofluorescence and ELISA
Serum anti-DSG1 and anti-DSG3 antibody titers can be measured by ELISA. Titers correlate with disease activity and are used to monitor response to treatment and guide tapering. Rising titers may predict clinical relapse before visible blistering recurs.
Pemphigus Variants
Pemphigus Foliaceus
Anti-DSG1 antibodies only (no anti-DSG3). Because DSG1 predominates in the upper epidermis and DSG3 compensates in mucosa, PF causes superficial skin crusting without mucosal involvement. The split is subcorneal (very superficial). Blisters are fragile and rarely intact; the typical presentation is honey-colored crusted erosions, often on the face and trunk. Less serious than PV but still autoimmune and requiring treatment.
Pemphigus Vegetans
A rare PV variant where erosions, particularly in intertriginous areas (axillae, groin), develop verrucous (wart-like), hypertrophic, vegetating plaques rather than simple erosions — thought to be a hyperplastic healing response.
Drug-Induced Pemphigus
Certain drugs can trigger a pemphigus-like syndrome: thiol-containing drugs (penicillamine, captopril) most commonly, but also ACE inhibitors, penicillins, cephalosporins, rifampin. Drug-induced PV may remit after drug withdrawal, though immunosuppression is often still needed acutely.
Induction Therapy: Corticosteroids
The goal of induction therapy is to rapidly suppress blister formation and achieve disease control. Before immunosuppressives, PV had 75% mortality; the introduction of systemic corticosteroids in the 1950s was transformative. Prednisone (or equivalent) remains the cornerstone of initial therapy.
- Starting dose: Prednisone 0.5–1.5 mg/kg/day (dose depends on disease severity). Most patients respond within 2–4 weeks with cessation of new blister formation.
- Duration: Maintained at induction dose until 80% healing, then tapered slowly (typically over months to years).
- Complications of prolonged corticosteroids: Osteoporosis, avascular necrosis, hyperglycemia/diabetes, adrenal suppression, hypertension, cataracts, infections, and weight gain. Prophylaxis with calcium, vitamin D, and bisphosphonates is routine. Corticosteroid morbidity historically exceeded disease morbidity in long-term survivors.
- Pulse steroids: Intravenous methylprednisolone pulses (1 g/day × 3 days) may be used for severe or rapidly progressing disease.
Adjuvant immunosuppressants are used to allow steroid tapering (steroid-sparing effect) and reduce cumulative steroid exposure.
Rituximab: The Modern Standard
Rituximab is an anti-CD20 monoclonal antibody that depletes B lymphocytes — the precursors to the plasma cells producing pathogenic anti-DSG IgG antibodies. It has fundamentally changed the treatment landscape for pemphigus vulgaris.
RITUX3 Trial
The landmark RITUX3 randomized controlled trial (Joly et al., 2017, Lancet) demonstrated that rituximab plus short-term prednisone outperformed long-term standard-dose prednisone for PV. At 24 months, 89% of rituximab patients were in complete remission off therapy versus 34% of prednisone-alone patients. Crucially, rituximab patients had significantly fewer corticosteroid-related adverse events. This trial established rituximab as the preferred first-line treatment for moderate-to-severe PV in many centers.
Rituximab Dosing in PV
- Rheumatoid arthritis protocol: 1,000 mg IV on days 0 and 14 (most commonly used for PV).
- Lymphoma protocol: 375 mg/m² weekly × 4 doses.
- Re-treatment at 6 months or at first sign of serologic relapse is common.
Why Rituximab Works So Well in PV
Unlike in rheumatoid arthritis (where many cytokines drive disease), pemphigus depends specifically and almost entirely on the production of pathogenic IgG antibodies. Depleting the B-cell compartment removes the antibody factory. Rituximab can achieve sustained complete remission in over 70–90% of patients, with some patients remaining in long-term remission after a single course.
Prior Steroid-Sparing Agents
Before rituximab availability, steroid-sparing was achieved with:
- Azathioprine: Antimetabolite, 1–3 mg/kg/day. Effective but slow onset (8–12 weeks). Requires TPMT enzyme testing before use.
- Mycophenolate mofetil (MMF): 2–3 g/day. Better tolerated than azathioprine, commonly used as adjuvant.
- Dapsone: For mild disease or pemphigus foliaceus.
- IV immunoglobulin (IVIg): Rapid effect through Fc receptor saturation and accelerated IgG catabolism; useful for refractory disease or perioperatively.
- Plasmapheresis: Rapid mechanical removal of circulating antibodies; short-lived effect, used as bridge therapy in severe acute flares.
Maintenance and Monitoring
After achieving disease control, maintenance focuses on preventing relapse while minimizing treatment toxicity.
- Steroid taper: Slow reduction — typically 10% of the current dose every 2–4 weeks once in remission. Premature tapering is the most common cause of relapse.
- DSG antibody monitoring: Anti-DSG3 and anti-DSG1 ELISA every 3–6 months. Rising titers predict relapse; may prompt preemptive rituximab re-dosing before blisters return.
- Skin care: Gentle handling, non-adhesive dressings for erosions, antiseptic washes to reduce secondary infection, ophthalmology review for ocular involvement.
- Vaccinations: Pneumococcal, influenza, and herpes zoster vaccines before or during immunosuppression (live vaccines contraindicated during active rituximab therapy).
- Oral care: Regular dental hygiene (gingival erosions may complicate dental procedures), oral rinses (dilute chlorhexidine), topical steroids for localized oral lesions.
Paraneoplastic Pemphigus
Paraneoplastic pemphigus (PNP) is a rare and severe variant triggered by an underlying neoplasm — most commonly non-Hodgkin lymphoma, chronic lymphocytic leukemia (CLL), Castleman disease, thymoma, or sarcoma. The antibodies in PNP target not only desmogleins but also plakins (envoplakin, periplakin, desmoplakin) and other structural proteins, producing a more devastating clinical picture.
PNP is distinguished from PV by:
- Severe, treatment-refractory stomatitis extending to the vermilion lip and esophagus
- Lichenoid skin lesions in addition to blistering
- Bronchiolitis obliterans (lung involvement) — causes life-threatening respiratory failure in some patients
- Multiprotein autoantibody pattern on immunoprecipitation
Prognosis of PNP is poor, largely determined by the underlying malignancy. Treating the cancer may improve PNP, but bronchiolitis obliterans tends to be irreversible. Overall mortality exceeds 90%.
Prognosis and Quality of Life
The prognosis of PV has been transformed over the past 70 years:
- Pre-corticosteroid era (before 1950): Mortality ~75% within 2–5 years
- Corticosteroid era (1950–2000): Mortality fell to ~10–15%, primarily from infections and steroid complications rather than the disease itself
- Rituximab era (post-2000): Mortality now <5%; most patients achieve complete remission
Even with successful treatment, PV significantly impacts quality of life:
- Oral erosions impair eating and speaking
- Widespread skin erosions cause pain, sleep disruption, and infection risk
- Prolonged immunosuppression increases infection risk (bacterial, viral, fungal) and long-term corticosteroid complications
- Psychological impact: depression and anxiety are common in chronic debilitating skin disease
- Flares and remissions create uncertainty; many patients require years of treatment
Approximately 30–40% of patients achieve durable complete remission off all therapy after rituximab treatment. The remainder require ongoing maintenance therapy or experience periodic relapses. Disease severity at diagnosis (number of body surface areas involved, oral severity, antibody titers) predicts long-term outcomes.
Research Papers
Key peer-reviewed studies on pemphigus vulgaris pathogenesis, diagnosis, and treatment. Each PMID link opens the study on PubMed.
- Joly P, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389(10083):2031-2040. PMID 28342637
- Stanley JR, Amagai M. Pemphigus, bullous impetigo, and the staphylococcal scalded-skin syndrome. N Engl J Med. 2006;355(17):1800-1810. PMID 17065642
- Amagai M, et al. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67(5):869-877. — Search PubMed
- Murrell DF, et al. Consensus statement on definitions of disease, end points, and therapeutic response for pemphigus. J Am Acad Dermatol. 2008;58(6):1043-1046. PMID 18339444
- Kasperkiewicz M, et al. Pemphigus. Nat Rev Dis Primers. 2017;3:17026. — Search PubMed
- Grando SA. Pemphigus autoimmunity: hypotheses and realities. Autoimmunity. 2012;45(1):7-35. — Search PubMed
- Ingen-Housz-Oro S, et al. First-line treatment of pemphigus vulgaris with a combination of rituximab and high-potency topical corticosteroids. JAMA Dermatol. 2018;154(3):348-350. PMID 29322186
- Amber KT, et al. An evidence-based review of the use of a humanized anti-CD20 monoclonal antibody, obinutuzumab, for inflammatory skin diseases. G Ital Dermatol Venereol. 2017;152(5):505-510. — Search PubMed
- Feliciani C, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172(4):867-877. — Search PubMed
- Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366(9479):61-73. — Search PubMed
- Werth VP. Clinical profile of autoimmune bullous diseases. Dermatol Clin. 2011;29(3):459-464. — Search PubMed
- Heelan K, et al. Pemphigus: the oral perspective. Oral Dis. 2015;21(4):e133-138. — Search PubMed
Curated PubMed topic searches:
- PubMed: Rituximab for PV
- PubMed: Desmoglein antibodies
- PubMed: DIF diagnosis
- PubMed: Corticosteroid therapy
- PubMed: Paraneoplastic PV
- PubMed: Oral mucosal PV
- PubMed: PV quality of life
- PubMed: Pemphigus foliaceus
Connections
- Dermatology
- Melanoma
- Psoriasis
- Eczema
- Vitiligo
- Contact Dermatitis
- Alopecia
- Lupus
- Rosacea
- Vitamin D3
- Seborrheic Dermatitis
- Oncology