Valine for Energy Metabolism
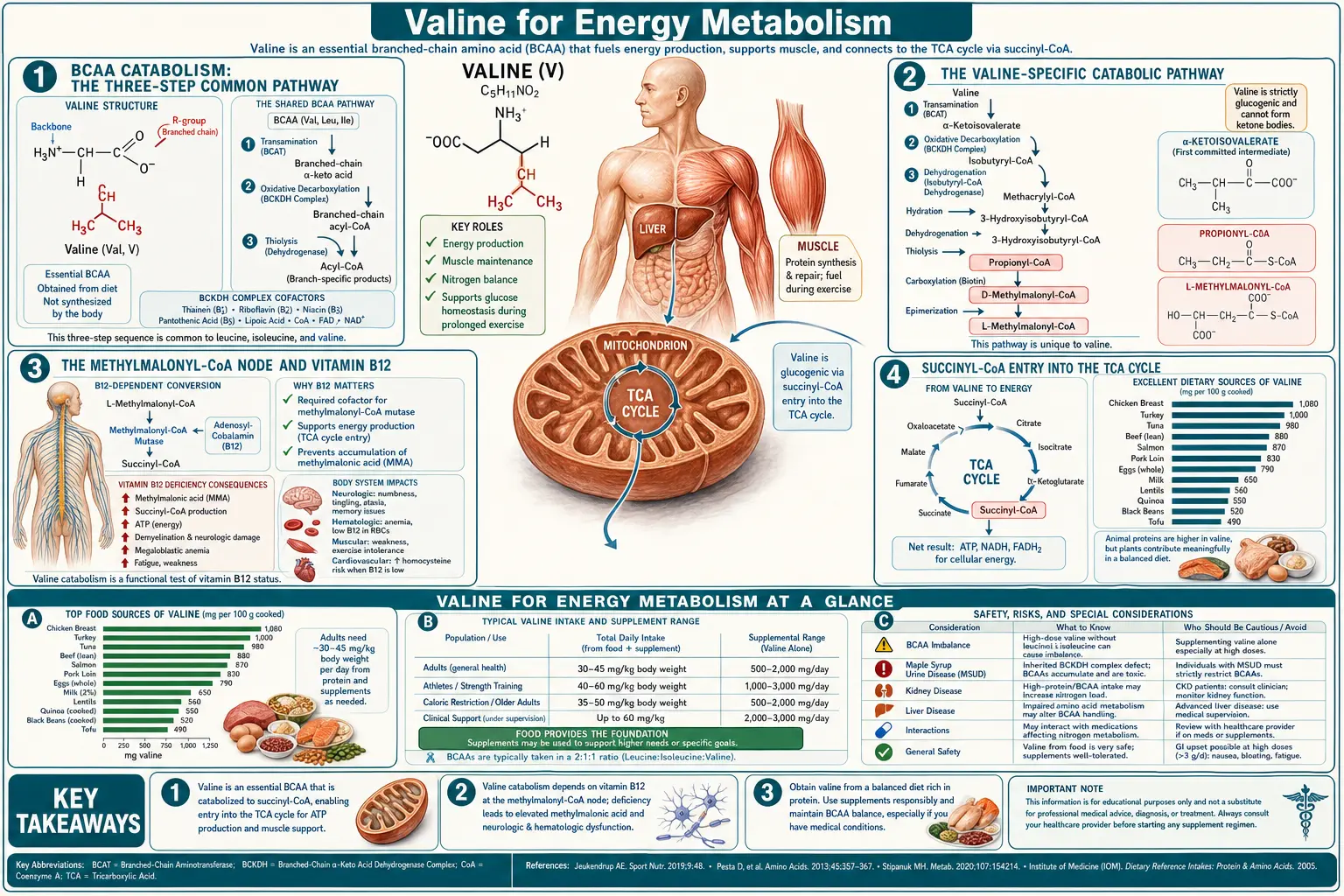
Valine is one of only a handful of amino acids that can feed carbon directly into the TCA (citric acid) cycle as an anaplerotic substrate — replenishing intermediates rather than just being burned for ATP. Its catabolic pathway runs through propionyl-CoA, methylmalonyl-CoA (the vitamin B12-dependent step), and succinyl-CoA, joining the TCA cycle at the succinate level. This is the same biochemical node that is disrupted in maple syrup urine disease (MSUD) — the classic genetic disorder of branched-chain alpha-ketoacid dehydrogenase (BCKAD) deficiency that elevates plasma BCAAs and their toxic ketoacids, producing the maple-syrup-scented urine and neurological crisis that gave the disease its name. A thiamine-responsive MSUD subtype reveals the critical role of vitamin B1 as the BCKAD cofactor. During fasting, valine liberated from muscle proteolysis becomes a gluconeogenic substrate, contributing alongside alanine and glutamine to maintain blood glucose for brain and red blood cells. This page maps that pathway end-to-end and explains its clinical implications.
Table of Contents
- BCAA Catabolism: The Three-Step Common Pathway
- The Valine-Specific Catabolic Pathway
- The Methylmalonyl-CoA Node and Vitamin B12
- Succinyl-CoA Entry into the TCA Cycle
- Valine as an Anaplerotic Substrate
- Maple Syrup Urine Disease (MSUD) and BCKAD Deficiency
- Thiamine-Responsive MSUD and Vitamin B1
- Gluconeogenesis from Muscle Proteolysis During Fasting
- Valine During Prolonged Exercise
- Regulation of BCKAD Activity
- Key Research Papers
- Connections
- Featured Videos
BCAA Catabolism: The Three-Step Common Pathway
All three branched-chain amino acids — leucine, isoleucine, and valine — share the first two steps of their catabolic pathway. Step one is reversible transamination by branched-chain aminotransferase (BCAT), which strips the alpha-amino group and transfers it to alpha-ketoglutarate to form glutamate, leaving behind a branched-chain alpha-ketoacid. For valine, the product is alpha-ketoisovalerate (KIV); for leucine, alpha-ketoisocaproate (KIC); for isoleucine, alpha-keto-beta-methylvalerate (KMV). Two BCAT isoforms exist: BCAT1 in the cytoplasm of many tissues (brain, kidney, ovary) and BCAT2 in mitochondria of essentially all tissues including skeletal muscle, the dominant site of BCAA transamination quantitatively.
Step two is the irreversible oxidative decarboxylation by the branched-chain alpha-ketoacid dehydrogenase complex (BCKAD or BCKDH), a mitochondrial multienzyme complex structurally and mechanistically analogous to pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase. BCKAD removes CO2 from the alpha-ketoacid and attaches the remaining carbon skeleton to coenzyme A as a branched-chain acyl-CoA. The complex requires five cofactors: thiamine pyrophosphate (TPP, from vitamin B1), lipoamide (from lipoic acid), coenzyme A (from pantothenic acid), FAD (from riboflavin), and NAD+ (from niacin). Deficiency of any of these B-vitamin cofactors can impair BCKAD activity, although the most clinically dramatic deficiency state is the genetic loss of function that causes maple syrup urine disease.
BCKAD is the rate-limiting enzyme of the entire BCAA catabolic pathway. It is heavily regulated by phosphorylation: BCKAD kinase phosphorylates and inactivates the complex; BCKAD phosphatase dephosphorylates and activates it. The kinase is itself inhibited by elevated levels of branched-chain alpha-ketoacids (substrate-level feedback) and is upregulated by glucagon and fasting. Net effect: in the fed state, BCKAD is largely inactive and dietary BCAAs are preserved for protein synthesis; in fasting or carbohydrate restriction, BCKAD activates and BCAAs flow into the TCA cycle as fuel.
From here, each BCAA diverges into its own product-specific catabolic steps. Leucine yields acetyl-CoA and acetoacetate (purely ketogenic). Isoleucine yields both acetyl-CoA and propionyl-CoA (mixed ketogenic and glucogenic). Valine yields propionyl-CoA only (purely glucogenic). The downstream paths from propionyl-CoA are what make valine unique among the BCAAs and what tie it to vitamin B12 metabolism.
The Valine-Specific Catabolic Pathway
After BCKAD action on alpha-ketoisovalerate, valine's carbon skeleton continues through the following sequence:
- Isobutyryl-CoA (the immediate BCKAD product from valine)
- Dehydrogenation by isobutyryl-CoA dehydrogenase to methacrylyl-CoA, with electrons donated to FADH2 and the electron transport chain
- Hydration by enoyl-CoA hydratase to 3-hydroxyisobutyryl-CoA
- Hydrolysis by 3-hydroxyisobutyryl-CoA hydrolase to 3-hydroxyisobutyrate (free acid form, which can leave the mitochondrion and circulate)
- Oxidation by 3-hydroxyisobutyrate dehydrogenase to methylmalonate semialdehyde
- Oxidation and CoA-attachment by methylmalonate semialdehyde dehydrogenase to propionyl-CoA
- Propionyl-CoA (the convergent node shared with isoleucine catabolism, odd-chain fatty acid oxidation, and methionine/threonine catabolism)
- Carboxylation by propionyl-CoA carboxylase (biotin-dependent) to D-methylmalonyl-CoA
- Isomerization by methylmalonyl-CoA racemase to L-methylmalonyl-CoA
- L-methylmalonyl-CoA isomerization by methylmalonyl-CoA mutase (vitamin B12-dependent) to succinyl-CoA
- Succinyl-CoA entry into the TCA cycle
The pathway has ten enzymatic steps from valine to succinyl-CoA, the largest number of any BCAA. Genetic defects in any of these enzymes produce specific organic acidemias, the most clinically important being the propionyl-CoA carboxylase defect (propionic acidemia) and the methylmalonyl-CoA mutase defect (methylmalonic acidemia). These are inborn errors of metabolism diagnosed by elevated organic acids in urine and treated by dietary BCAA restriction (especially valine, isoleucine, methionine, and threonine, all of which converge on propionyl-CoA).
The Methylmalonyl-CoA Node and Vitamin B12
The conversion of L-methylmalonyl-CoA to succinyl-CoA by methylmalonyl-CoA mutase is one of only two reactions in human biochemistry that require vitamin B12 (cobalamin) as cofactor — the other being the homocysteine-to-methionine conversion by methionine synthase. The B12 form required by methylmalonyl-CoA mutase is 5'-deoxyadenosylcobalamin (AdoCbl), an organometallic compound with a covalent cobalt-carbon bond unique in mammalian biochemistry.
Functional B12 deficiency — whether from inadequate intake (strict veganism without supplementation), malabsorption (atrophic gastritis, pernicious anemia, ileal disease, metformin use), or genetic disorders of B12 transport and processing — causes a buildup of methylmalonyl-CoA, which hydrolyzes spontaneously to methylmalonic acid (MMA) and accumulates in plasma and urine. Serum or urinary methylmalonic acid is therefore the single most sensitive biochemical marker of functional B12 deficiency, more sensitive than serum B12 itself (which can be in the normal range despite tissue-level deficiency). MMA testing is the standard follow-up for any patient with borderline B12 and suspected neurological or hematological B12 deficiency.
The methylmalonyl-CoA buildup has consequences beyond just its diagnostic utility. Methylmalonyl-CoA is a structural analog of malonyl-CoA, the regulatory and synthetic precursor for fatty acid synthesis. Elevated methylmalonyl-CoA inhibits malonyl-CoA-dependent processes and is thought to contribute to the abnormal lipid composition (odd-chain and branched-chain fatty acids) seen in untreated B12 deficiency and in inborn errors of methylmalonic acidemia. This abnormal lipid synthesis affects myelin composition and is one proposed mechanism for the subacute combined degeneration of the dorsal columns and corticospinal tracts that characterizes severe B12 deficiency neurologically.
The valine connection means that any patient with elevated MMA — whether from B12 deficiency or from genetic methylmalonic acidemia — has a valine-restricted dietary recommendation as part of management. In genetic methylmalonic acidemia, the valine restriction is strict and lifelong; in B12 deficiency, MMA normalizes with B12 repletion and no dietary restriction is needed.
Succinyl-CoA Entry into the TCA Cycle
Succinyl-CoA is one of eight TCA cycle intermediates and one of the most metabolically versatile. Once formed from valine catabolism, succinyl-CoA can be processed through several alternative routes:
- TCA cycle progression: succinyl-CoA synthetase converts succinyl-CoA to succinate, generating one GTP (substrate-level phosphorylation). Succinate then proceeds through fumarate, malate, oxaloacetate, and back to citrate — one full turn of the cycle producing 1 GTP, 2 NADH, and 1 FADH2 (yielding approximately 9-10 ATP per turn through oxidative phosphorylation).
- Heme synthesis: succinyl-CoA condenses with glycine in the mitochondrion (catalyzed by ALA synthase, the rate-limiting and B6-dependent enzyme of heme synthesis) to form delta-aminolevulinate (ALA), the first committed precursor of heme. A measurable fraction of valine-derived succinyl-CoA contributes to heme synthesis, particularly in erythroid precursors in the bone marrow.
- Gluconeogenesis: in the liver, succinate from the TCA cycle can be drawn off as malate, exported from the mitochondrion, converted to oxaloacetate in the cytoplasm, and channeled into gluconeogenesis via phosphoenolpyruvate carboxykinase (PEPCK). This is the route by which valine ultimately contributes to blood glucose during fasting.
- Anaplerotic replenishment: when TCA intermediates are pulled out for biosynthesis (heme, glutamate, aspartate, gluconeogenesis), they must be replenished or the cycle stalls. Succinyl-CoA from valine catabolism is one of several key anaplerotic inputs that keep the cycle topped up.
The succinyl-CoA entry point also explains why valine is mildly insulinogenic. Succinyl-CoA and its downstream TCA intermediates produce ATP, which closes ATP-sensitive potassium channels in pancreatic beta-cells and triggers insulin secretion. This is a small effect compared to glucose or to direct amino-acid-stimulated insulin release (which is most pronounced for leucine), but it contributes to the integrated metabolic response to a protein-containing meal.
Valine as an Anaplerotic Substrate
Anaplerosis — the replenishment of TCA cycle intermediates — is a biochemical concept that often gets short shrift in introductory metabolism teaching, but it is crucial for understanding why some amino acids are particularly valuable in catabolic and high-flux states. The TCA cycle is not a closed loop in the steady state; intermediates are continuously drawn off for biosynthesis (aspartate from oxaloacetate, glutamate from alpha-ketoglutarate, heme from succinyl-CoA), and if they are not replenished, cycle flux drops and aerobic ATP production collapses.
The principal anaplerotic enzyme is pyruvate carboxylase, which converts pyruvate to oxaloacetate (biotin-dependent), and the principal anaplerotic substrates are pyruvate (from glucose), glutamine (from circulating amino acid pool), and the propionyl-CoA-yielding amino acids: valine, isoleucine, methionine, and threonine. Of these, valine and isoleucine quantitatively dominate during fasting, exercise, and any state where dietary protein is being catabolized.
The clinical significance: in conditions where TCA flux is impaired by intermediate depletion — some forms of mitochondrial disease, severe sepsis with mitochondrial dysfunction, certain organic acidemias — valine and other anaplerotic substrates become limiting for energy production. Triheptanoin (a medium-odd-chain triglyceride that yields propionyl-CoA upon oxidation) has been developed as a therapy for long-chain fatty acid oxidation disorders specifically because it can refill the TCA cycle through the same propionyl-CoA-to-succinyl-CoA pathway that valine uses. The biochemistry of valine catabolism is therefore directly applicable to a small but important set of inherited metabolic disorders.
Maple Syrup Urine Disease (MSUD) and BCKAD Deficiency
Maple syrup urine disease, first described in 1954 by Menkes, Hurst, and Craig, is the prototypical disorder of BCAA catabolism. It is caused by autosomal recessive mutations in any of the four subunits of the BCKAD complex: E1alpha (BCKDHA gene), E1beta (BCKDHB gene), E2 (DBT gene), or E3 (DLD gene; this last subunit is shared with pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase, so DLD mutations produce a more severe combined phenotype).
With BCKAD nonfunctional, all three BCAAs and their alpha-ketoacids accumulate to dramatic levels in plasma, urine, and CSF. The disease name comes from the distinctive sweet, maple-syrup-like odor of the urine, produced by the sotolone-like aroma of accumulated branched-chain ketoacids (especially the leucine-derived alpha-ketoisocaproate). In the classic neonatal form, infants appear normal at birth but within days of beginning protein feeds develop progressive lethargy, feeding intolerance, alternating hyper- and hypotonia, opisthotonos, and ultimately seizures, coma, and cerebral edema if untreated. Mortality without treatment is essentially 100% in the first month of life.
The mechanism of neurotoxicity is multifactorial:
- Elevated plasma leucine competes with other large neutral amino acids (LNAAs) for transport across the blood-brain barrier via LAT1, depleting brain levels of phenylalanine, tyrosine, tryptophan, methionine, histidine, and threonine
- The neurotransmitter precursors tyrosine and tryptophan become brain-deficient, impairing catecholamine and serotonin synthesis
- Branched-chain alpha-ketoacids are directly neurotoxic, particularly alpha-ketoisocaproate, which inhibits brain alpha-ketoglutarate dehydrogenase and disrupts brain energy metabolism
- Severe accumulation causes cerebral edema, the leading cause of acute mortality in untreated metabolic crisis
Treatment is lifelong dietary restriction of BCAAs (especially leucine, which is the most neurotoxic), using specialized BCAA-free amino acid formulas to provide other essential amino acids, careful protein management, and emergency hospitalization for any catabolic illness or trauma that mobilizes endogenous BCAAs. Some patients are candidates for liver transplantation, which provides functional BCKAD activity systemically and allows return to a normal diet.
Thiamine-Responsive MSUD and Vitamin B1
A minority of MSUD patients — approximately 2-5% — have a milder form of the disease that responds dramatically to high-dose pharmacologic vitamin B1 (thiamine) supplementation. These patients carry mutations in the E1alpha or E2 subunits of BCKAD that destabilize the complex's binding of its thiamine pyrophosphate cofactor. Saturating the residual enzyme with high concentrations of TPP partially restores activity, lowering plasma BCAA levels and reducing the dietary restriction requirements.
Thiamine-responsive MSUD is treated with 10-200 mg/day of oral thiamine (compared to the 1-2 mg/day RDA for healthy adults), typically alongside continued but less restrictive dietary BCAA limitation. The dramatic response in these patients — plasma leucine levels can drop by 50-80% within days of starting high-dose thiamine — demonstrates the principle that cofactor availability can rescue residual enzyme function in enzymopathies with partial activity remaining.
The broader implication of thiamine-responsive MSUD extends to wild-type BCKAD activity in the general population. Thiamine deficiency — chronic alcoholism, post-bariatric surgery, prolonged severe malnutrition, hyperemesis gravidarum — impairs BCKAD function and can elevate plasma BCAAs and their ketoacids, though usually not to MSUD-like levels. This is one of several biochemical mechanisms by which thiamine deficiency produces neurological symptoms (Wernicke encephalopathy, beriberi). The thiamine RDA is built on the assumption of adequate intake; tissues with high TCA flux (brain, heart, skeletal muscle in athletes) may have higher functional requirements.
Thiamine status is most reliably assessed by erythrocyte transketolase activity with and without TPP addition (the "TPP effect" — a percentage increase above 15% suggests deficiency), or by whole-blood thiamine pyrophosphate measurement. Serum thiamine is a poor marker because most thiamine circulates intracellularly. For at-risk populations (chronic alcoholism, post-bariatric, hyperemesis gravidarum), empiric thiamine supplementation at 100 mg/day oral or 100-500 mg IV for acute presentation is standard of care.
Gluconeogenesis from Muscle Proteolysis During Fasting
During an overnight fast, plasma glucose is maintained primarily by hepatic glycogenolysis (the breakdown of stored liver glycogen). As liver glycogen depletes — typically within 12-18 hours of fasting — gluconeogenesis takes over as the dominant glucose source. The substrates for gluconeogenesis are: lactate (from anaerobic glycolysis, especially in red blood cells and exercising muscle), glycerol (from triglyceride lipolysis), and glucogenic amino acids (from net muscle protein breakdown).
Among the amino acids, the principal gluconeogenic substrates released from muscle are alanine and glutamine, accounting for approximately 50-60% of the amino acid nitrogen exported from skeletal muscle during fasting. This is the basis of the glucose-alanine cycle: muscle proteolysis liberates BCAAs, which donate their amino groups (via BCAT transamination) to pyruvate, forming alanine; alanine is exported to the liver and reconverted to pyruvate (and then to glucose), with the amino group going into the urea cycle. The carbon skeleton of valine (and the other BCAAs) is largely retained in the muscle, where it enters the TCA cycle as succinyl-CoA and is oxidized for local ATP.
However, a measurable fraction of valine carbon does reach the liver and contribute to gluconeogenesis directly. The succinyl-CoA from valine can be exported as malate, converted in the liver cytoplasm to oxaloacetate and then to phosphoenolpyruvate, and finally to glucose. The quantitative contribution of valine carbon to glucose output during fasting is on the order of 5-10% of total gluconeogenesis — modest but not negligible.
This biochemistry explains why valine (and BCAAs more broadly) are not classically considered "sparing" of muscle during fasting in the same way that adequate carbohydrate intake is. The amino nitrogen is preserved through transamination to alanine, but the carbon skeleton of valine derived from muscle proteolysis is being oxidized as fuel and lost to the protein pool. Sustained fasting therefore depletes muscle protein even with adequate metabolic adaptation, and prolonged fasting (beyond ~3 weeks) ultimately compromises diaphragm and cardiac muscle function in a way that becomes life-threatening — the historical end point of hunger strikes and famine.
Valine During Prolonged Exercise
During sustained aerobic exercise, fuel selection shifts progressively. The first 30 minutes draw heavily on muscle glycogen and circulating glucose; as glycogen depletes (typically after 60-90 minutes of moderate-to-high-intensity exercise), free fatty acids become the dominant fuel; in very prolonged exercise (4+ hours), amino acid oxidation can account for 5-10% of total energy expenditure. Valine and isoleucine are the principal amino acids oxidized in this context, because of their direct entry into the TCA cycle (and because BCKAD activity in exercising muscle increases approximately 3-5 fold compared to rest).
The practical consequence for endurance athletes is twofold. First, prolonged exercise produces measurable muscle protein breakdown, with elevated urinary 3-methylhistidine (a marker of myofibrillar protein degradation) for 24-48 hours after a very long event. Second, recovery from prolonged exercise requires adequate protein intake to rebuild what was catabolized. Contemporary sports nutrition recommendations for endurance athletes are 1.2-1.6 g/kg/day total protein, with up to 1.8 g/kg/day for ultra-endurance and stage-race contexts.
Intra-workout BCAA supplementation has been marketed to endurance athletes on the rationale that providing exogenous BCAAs reduces the need for endogenous muscle proteolysis. Controlled trials have shown small effects: a 2017 meta-analysis of intra-workout BCAA in endurance contexts found a modest reduction in perceived exertion (~5-8%) and a small reduction in markers of muscle damage, but no clear effect on actual endurance performance or recovery beyond what total protein intake provides. The probable mechanism of the perceived-exertion benefit is the central fatigue hypothesis discussed on the cognitive performance page — BCAA intake competes with tryptophan for brain entry and reduces serotonin-mediated central fatigue signaling.
Regulation of BCKAD Activity
BCKAD is one of the most heavily regulated metabolic enzymes in the body, reflecting the importance of conserving BCAA stores in the fed state while permitting rapid catabolism in fasting or exercise. Key regulatory inputs include:
- Phosphorylation state — BCKAD kinase phosphorylates and inactivates the complex; BCKAD phosphatase reverses this. The kinase is the dominant regulator in vivo; pharmacological inhibition of BCKAD kinase by clofibric acid analogs (currently in research stages) activates the complex and lowers plasma BCAAs.
- Substrate-level feedback — elevated branched-chain alpha-ketoacids (the BCKAD substrates) inhibit the kinase and thereby activate the complex, providing autoregulatory throughput control.
- Hormonal control — insulin suppresses BCKAD activity (conserves BCAA for protein synthesis in the fed state); glucagon and glucocorticoids activate BCKAD (mobilize BCAA for fuel in fasting and stress).
- Tissue-specific expression — BCKAD activity is highest in liver and kidney, intermediate in heart and skeletal muscle, lower in adipose tissue and brain. The relative tissue distribution shifts the flux of BCAA catabolism between organs depending on the metabolic state.
- Inter-organ shuttling — in the fed state, the liver oxidizes a small fraction of dietary BCAAs and releases most into circulation for uptake by muscle. In the fasted or exercising state, muscle BCAA proteolysis releases BCAA and BCAA-derived alanine to the liver for gluconeogenesis.
Recent research has implicated dysregulated BCAA catabolism in metabolic disease. Elevated plasma BCAAs (and their ketoacids) are among the strongest amino acid biomarkers of insulin resistance and type 2 diabetes risk, observed years before clinical diabetes onset in longitudinal studies. The mechanism is debated — whether elevated BCAAs cause insulin resistance, or insulin resistance impairs BCKAD activity and elevates BCAAs secondarily — but the association is robust across populations. This has stimulated interest in BCKAD activators as a potential therapeutic strategy for type 2 diabetes prevention.
Key Research Papers
- Menkes JH, Hurst PL, Craig JM (1954). A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urinary substance. Pediatrics. — PubMed
- Strauss KA, Puffenberger EG, Morton DH (2020). Maple syrup urine disease. GeneReviews. — PubMed
- Harper AE, Miller RH, Block KP (1984). Branched-chain amino acid metabolism. Annual Review of Nutrition. — PubMed
- Chuang DT et al. (2006). Maple syrup urine disease (branched-chain ketoaciduria). In: Scriver's Online Metabolic and Molecular Bases of Inherited Disease. — PubMed
- Wang TJ et al. (2011). Metabolite profiles and the risk of developing diabetes. Nature Medicine. — PubMed
- Newgard CB et al. (2009). A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metabolism. — PubMed
- Lynch CJ, Adams SH (2014). Branched-chain amino acids in metabolic signalling and insulin resistance. Nature Reviews Endocrinology. — PubMed
- Brunetti-Pierri N et al. (2011). Phenylbutyrate therapy for maple syrup urine disease. Human Molecular Genetics. — PubMed
- Mazariegos GV et al. (2012). Liver transplantation for classical maple syrup urine disease. Journal of Pediatrics. — PubMed
- Roe CR, Brunengraber H (2015). Anaplerotic treatment of long-chain fat oxidation disorders with triheptanoin. Molecular Genetics and Metabolism. — PubMed
- Banerjee R, Ragsdale SW (2003). The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annual Review of Biochemistry. — PubMed
- Stabler SP (2013). Vitamin B12 deficiency. NEJM. — PubMed
PubMed Topic Searches
- PubMed: BCKAD regulation
- PubMed: MSUD treatment
- PubMed: Thiamine-responsive MSUD
- PubMed: MMA and B12 status
- PubMed: BCAA and insulin resistance
Connections
- Valine Overview
- Valine Benefits Hub
- Valine for Muscle Protein Synthesis
- Valine for Cognitive Performance
- Valine for Nitrogen Balance & Wound Healing
- Leucine (BCAA Partner)
- Isoleucine (BCAA Partner)
- Alanine (Glucose-Alanine Cycle)
- Glutamine
- Vitamin B12 (Methylmalonyl-CoA Mutase Cofactor)
- Vitamin B1 / Thiamine (BCKAD Cofactor)
- Vitamin B2 / Riboflavin
- Insulin Resistance
- Fasting
- All Amino Acids