Membranous Nephropathy
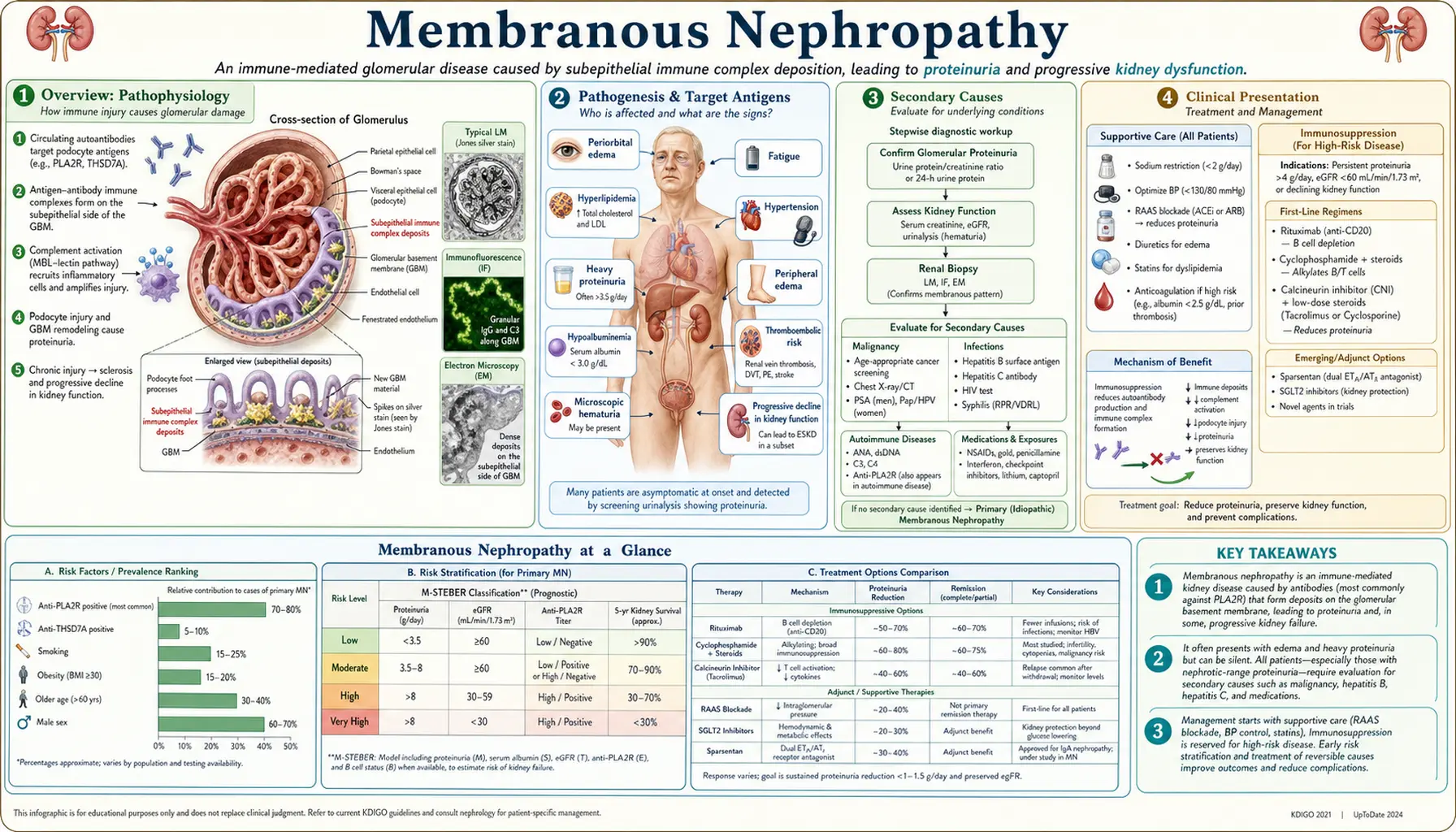
Membranous nephropathy (MN) is the most common cause of primary nephrotic syndrome in white adults over 40. It is an autoimmune glomerular disease in which antibodies — most often against M-type phospholipase A2 receptor (PLA2R1) on podocytes — form subepithelial immune complex deposits along the outer aspect of the glomerular basement membrane. Complement activation via C5b-9 (membrane attack complex) damages podocytes, producing massive proteinuria, edema, hypoalbuminemia, hyperlipidemia, and a heightened risk of venous thromboembolism including renal vein thrombosis.
Table of Contents
- Overview
- Pathogenesis and Target Antigens
- Secondary Causes
- Clinical Presentation
- Diagnosis
- Histopathology
- Staging and Risk Stratification
- Treatment
- Complications
- Prognosis and the Rule of Thirds
- Research Papers (PubMed searches)
- References
- Featured Videos
1. Overview
Membranous nephropathy (MN), also called membranous glomerulonephritis (MGN), is a glomerular disease defined by subepithelial immune complex deposition along the outer surface of the glomerular basement membrane (GBM). These deposits are formed in situ: circulating antibodies bind antigens expressed on podocyte foot processes, activating the complement cascade and generating the C5b-9 membrane attack complex, which injures podocytes and leads to disruption of the filtration barrier, proteinuria, and eventual GBM thickening.
MN accounts for approximately 20–30% of adult nephrotic syndrome cases in white populations, making it the leading primary cause in adults over 40. It is rarer in children and in populations of African descent, where FSGS predominates. The discovery of anti-PLA2R antibodies in 2009 transformed MN from a purely biopsy-defined entity to a serologically diagnosable autoimmune disease, enabling non-invasive diagnosis, disease activity monitoring, and targeted therapy.
Primary (idiopathic) MN accounts for 70–80% of cases; the remainder are secondary to malignancy, systemic autoimmune disease, chronic infections, or drugs. Distinguishing primary from secondary MN is critical because treatment differs fundamentally.
2. Pathogenesis and Target Antigens
The central event in MN is the formation of immune complexes on the subepithelial surface of the GBM (the side facing podocyte foot processes, outside the glomerular capillary). Circulating IgG antibodies bind antigens expressed on podocytes; the resulting immune complexes activate the complement cascade locally. C5b-9 inserts into the podocyte membrane, causing sublytic injury that disrupts slit-diaphragm proteins, foot-process cytoskeletal architecture, and the charge-selective filtration barrier.
Anti-PLA2R1 antibody: The dominant autoantigen in primary MN. M-type phospholipase A2 receptor (PLA2R1) is a 185 kDa transmembrane receptor expressed abundantly on the apical surface of glomerular podocytes. Circulating anti-PLA2R IgG4 antibodies (predominantly IgG4 subclass, which fixes complement poorly via classical pathway but activates it via the lectin pathway) are found in 70–80% of primary MN cases. Antibody titers correlate with disease activity: rising titers signal relapse; falling titers (immunological remission) typically precede proteinuria remission by weeks to months. Serum anti-PLA2R testing (ELISA or indirect immunofluorescence on HEK293 cells transfected with PLA2R) has sensitivity ~75% and specificity >99% for primary MN. Renal biopsy immunofluorescence staining for PLA2R antigen in deposits increases sensitivity to ~85%.
Anti-THSD7A antibody: Thrombospondin type-1 domain–containing 7A, found in 3–5% of primary MN and associated with thyroid malignancy and recurrence post-transplant. Titers are also monitored in anti-THSD7A-positive patients.
Anti-NELL-1 (neural EGFL-like 1): Identified in ~5–10% of PLA2R-negative primary MN. Higher frequency in older patients.
Other emerging antigens: SEMA3B (semaphorin 3B), NCAM1, EXT1/EXT2 (associated with lupus-associated MN), PCDH7, and CFL2. Multiplex antigen testing is increasingly available commercially and may replace the sequential single-antigen approach.
In secondary MN, planted exogenous antigens (hepatitis B e antigen, tumor antigens, drug haptens) or circulating immune complexes from autoimmune disease (lupus) deposit subepithelially by different mechanisms.
3. Secondary Causes
Secondary MN must be excluded in every patient before diagnosis of primary (idiopathic) MN. Secondary causes account for 20–30% of MN and require treatment of the underlying condition, not immunosuppression for glomerulonephritis.
Malignancy-associated MN: The most clinically important secondary cause in adults over 60. Carcinomas (colon, lung, breast, gastric, renal) and lymphoma, particularly non-Hodgkin lymphoma. Anti-PLA2R antibodies are usually absent in malignancy-associated MN (though exceptions occur). Anti-THSD7A is associated with thyroid malignancy. Any PLA2R-negative MN in a patient over 60 should prompt malignancy screening: CT chest/abdomen/pelvis, age-appropriate cancer screens, and PET-CT if clinical suspicion is high. Treating the malignancy often leads to MN remission.
Systemic lupus erythematosus (SLE): ISN/RPS Class V lupus nephritis is the lupus-specific form of MN. Characterized histologically by EXT1/EXT2 antigen deposits, IgG1–IgG3 (not IgG4) in GBM, “full house” immunofluorescence (IgG, IgM, IgA, C3, C1q), mesangial immune complex deposits, and subendothelial deposits. These features distinguish lupus MN from primary MN. Serological workup: ANA, anti-dsDNA, anti-Sm, complement C3/C4.
Hepatitis B–associated MN: Particularly in regions of Asia where hepatitis B is endemic. Hepatitis B e antigen (HBeAg) is deposited in the GBM. Testing: HBsAg, anti-HBs, HBeAg, anti-HBe, HBV DNA. Treatment: antiviral therapy (tenofovir or entecavir) leads to MN remission as viral suppression occurs. Immunosuppression alone is not recommended.
Hepatitis C: More commonly causes cryoglobulinemic membranoproliferative GN than MN, but MN has been reported.
Drug-induced MN: Classic agents include NSAIDs (particularly in elderly), penicillamine, gold salts (used in rheumatoid arthritis), bucillamine, and captopril. More recently: checkpoint inhibitors (PD-1/PD-L1 inhibitors can unmask autoimmune MN). Cessation of the offending drug often leads to partial or complete remission.
Sarcoidosis and other granulomatous diseases are rare secondary causes.
Standard secondary workup panel: ANA, anti-dsDNA, C3, C4, HBsAg, anti-HCV antibody, serum protein electrophoresis (paraprotein), CT chest/abdomen/pelvis (malignancy screen), anti-PLA2R and anti-THSD7A antibodies (positive = favors primary).
4. Clinical Presentation
MN most commonly presents in adults aged 40–70 years (peak incidence in the 6th decade); it is uncommon in children. Males are affected approximately twice as frequently as females in primary MN.
Nephrotic syndrome is the presenting feature in ~80% of patients:
- Proteinuria >3.5 g/day (often 5–15 g/day; frank nephrotic-range): frothy or foamy urine, typically non-selective (albumin + IgG + transferrin loss)
- Edema: peripheral (lower limb, periorbital worse in morning), ascites, pleural effusions in severe cases; caused by hypoalbuminemia and sodium retention
- Hypoalbuminemia (<3.5 g/dL; often <2.0 g/dL in severe disease)
- Hyperlipidemia: elevated LDL-cholesterol, triglycerides, total cholesterol; hepatic lipoprotein synthesis upregulated in response to oncotic pressure loss
- Lipiduria: oval fat bodies, fatty casts, and Maltese crosses under polarized light on urine microscopy
Microscopic hematuria is present in 20–40% of cases; frank gross hematuria is rare and should raise suspicion for superimposed crescentic GN or malignancy.
Hypertension is present in 30–40% at presentation; increases in frequency with disease progression and CKD.
Thromboembolic complications: MN carries the highest VTE risk of all nephrotic syndromes. Renal vein thrombosis occurs in 25–50% of MN patients with severe nephrotic syndrome (hypoalbuminemia <2.0 g/dL). Mechanism: urinary losses of anticoagulant proteins (antithrombin III, protein C, protein S) combined with elevated hepatic synthesis of procoagulant factors (fibrinogen, factors V and VIII). Deep vein thrombosis and pulmonary embolism are also common. Flank pain, sudden worsening proteinuria, or new hematuria may signal renal vein thrombosis.
Subclinical presentation: ~20% are detected incidentally on urinalysis (proteinuria without overt edema).
5. Diagnosis
The diagnostic approach integrates serology, renal biopsy (when indicated), and exclusion of secondary causes.
Serum anti-PLA2R antibody: ELISA or immunofluorescence testing. A positive titer in a patient with nephrotic syndrome is virtually diagnostic of primary MN without renal biopsy, particularly if secondary causes are excluded. Sensitivity ~75%, specificity >99%. Some centers manage seropositive patients (especially elderly, high surgical risk) without biopsy, reserving biopsy for seronegative cases or diagnostic uncertainty. Titers are used to monitor disease activity and guide treatment decisions: titer declining after rituximab therapy indicates immunological response even before proteinuria falls.
Anti-THSD7A antibody: Tested if anti-PLA2R is negative; guides malignancy workup (thyroid) and predicts post-transplant recurrence.
Kidney biopsy: Remains the gold standard for diagnosis, especially in seronegative cases, suspected secondary MN, or when concurrent disease is suspected.
- Light microscopy (LM): Diffuse thickening of the GBM without significant hypercellularity (mesangial or endocapillary proliferation suggests secondary MN or concurrent disease). Silver stain (Jones methenamine silver) shows characteristic spikes projecting from the outer GBM surface — these are GBM matrix laid down between subepithelial immune complex deposits; later the deposits become incorporated and the GBM shows holes or “Swiss cheese” appearance.
- Immunofluorescence (IF): Granular IgG + C3 deposits along the GBM in a “capillary loop” pattern. In primary MN: predominantly IgG4 (with minor IgG1). In lupus MN: IgG1–IgG3 dominant, full-house (IgG+IgM+IgA+C3+C1q). PLA2R antigen co-localizes with IgG4 deposits in primary MN — PLA2R staining of biopsy has higher sensitivity than serology alone.
- Electron microscopy (EM): Subepithelial electron-dense deposits (Ehrenreich–Churg staging: Stage I — small scattered deposits, normal GBM thickness; Stage II — deposits with early spikes; Stage III — deposits encircled by spikes/GBM matrix, significant thickening; Stage IV — deposits fully incorporated, markedly irregular GBM, possibly sclerosis). Foot process effacement is global. Subendothelial or mesangial deposits suggest secondary causes (lupus, membranoproliferative GN).
Urine studies: Spot urine protein-to-creatinine ratio or 24-hour urine protein; urine microscopy (fat bodies, casts); urine protein electrophoresis if selective proteinuria suspected.
Laboratory workup: Serum albumin, lipid panel, eGFR, ANA, anti-dsDNA, C3, C4, HBsAg, anti-HCV, CBC, and serum protein electrophoresis. CT chest/abdomen/pelvis for malignancy screening, especially in patients over 60 with PLA2R-negative biopsy.
6. Histopathology
The morphological hallmark of membranous nephropathy is diffuse subepithelial immune complex deposition producing characteristic GBM alterations. The staging system devised by Ehrenreich and Churg (1968) describes four sequential stages based on light and electron microscopy.
Stage I (Early): Small, sparse subepithelial deposits visible only on EM. GBM thickness is normal or only minimally increased. Silver stain and periodic acid–Schiff (PAS) stain appear normal or show very subtle GBM changes. IF shows faint granular capillary loop IgG. Diagnosis may be missed on LM alone at this stage.
Stage II (Established): EM shows moderate-sized subepithelial deposits. Silver stain demonstrates irregular projections (“spikes”) of GBM material extending outward between deposits. This spike-and-dome pattern is pathognomonic when seen on silver stain. IF is strongly positive. GBM is diffusely thickened.
Stage III (Advanced): Deposits are now surrounded by and incorporated into GBM matrix (dome-and-spike advances to fully encircled deposits). GBM shows marked, irregular thickening. On silver stain, encircled deposits create a “Swiss cheese” or “bubble” pattern of lucencies within the thickened GBM. Foot-process effacement is extensive. Podocyte vacuolization and microvillous transformation of foot processes are present.
Stage IV (Sclerotic): Old, electron-lucent (dissolved) deposits leave vacuoles within a markedly thickened and irregular GBM. Glomerulosclerosis begins to appear. Tubular atrophy and interstitial fibrosis reflect chronic progressive injury. Stage IV correlates with irreversible nephron loss and diminished response to therapy.
Immunofluorescence antigen staining: Staining biopsy sections with anti-PLA2R antibody identifies PLA2R antigen within deposits, confirming primary MN even in seronegative patients (sensitivity ~85%). Absence of PLA2R and THSD7A in deposits strongly favors secondary MN and should prompt systematic secondary workup.
7. Staging and Risk Stratification
The natural history of untreated primary MN follows the Rule of Thirds: approximately one-third of patients achieve spontaneous complete remission, one-third achieve partial remission with persistent proteinuria, and one-third progress to ESRD over 10–15 years. This spontaneous remission rate justifies an initial period of conservative management in low-risk patients.
Risk stratification guides treatment timing:
Low risk: Proteinuria <3.5 g/day (or <4 g/day); stable eGFR; serum albumin >3.0 g/dL; anti-PLA2R titer low or undetectable. These patients are managed conservatively; 50% will remit spontaneously within 2 years. Active monitoring without immunosuppression is recommended.
Moderate risk: Proteinuria 3.5–8 g/day; preserved eGFR; no severe complications. Conservative management for 6 months; if no improvement, immunosuppression is initiated.
High risk: Proteinuria >8 g/day persistently; declining eGFR; severe hypoalbuminemia (<2.5 g/dL); life-threatening complications (thromboembolism, anasarca); high or rising anti-PLA2R titer. Immunosuppressive therapy is initiated promptly.
Additional prognostic markers: Male sex, older age, hypertension, and interstitial fibrosis on biopsy are associated with higher progression risk. Anti-PLA2R titer decline after rituximab is the strongest single predictor of proteinuria remission.
8. Treatment
Supportive Care (All Patients)
RAAS blockade: ACE inhibitors or ARBs for all patients with proteinuria regardless of blood pressure; target blood pressure <125/75 mmHg. Reduce proteinuria 30–50%, improve outcomes, and are renoprotective. Dual RAAS blockade (ACEi + ARB) carries significant adverse effects and is not routinely recommended.
Dietary sodium restriction: <2 g/day to reduce edema; loop diuretics (furosemide) for symptomatic edema.
Statin therapy: Indicated for MN-associated hyperlipidemia (cardiovascular risk reduction; at minimum moderate-intensity statin).
Anticoagulation: Prophylactic anticoagulation (warfarin or LMWH) is strongly considered when serum albumin <2.5 g/dL (and absent contraindications). Therapeutic anticoagulation for documented renal vein thrombosis or DVT/PE; typically 6–12 months or until nephrotic remission (whichever longer). Direct oral anticoagulants (DOACs) are used increasingly but data specific to MN is limited.
Immunosuppressive Therapy
Rituximab (first-line for immunosuppression): Anti-CD20 monoclonal antibody depletes B cells, suppressing antibody production. The MENTOR trial (Fervenza et al., 2019) randomized patients to rituximab vs cyclosporine; rituximab achieved superior complete or partial remission at 24 months (60% vs 20% complete remission) with fewer relapses and a better safety profile. Rituximab is now recommended as first-line immunosuppressive therapy by KDIGO 2021 guidelines for moderate- and high-risk primary MN. Dosing: 375 mg/m² ×4 weekly doses, or 1 g ×2 doses 14 days apart (MENTOR protocol). Anti-PLA2R titers are monitored at 3–6 months post-treatment; falling titers indicate immunological response. Proteinuria remission may lag immunological remission by 3–12 months.
Cyclophosphamide-based regimens (Ponticelli regimen): Alternating monthly steroids and cyclophosphamide (Ponticelli’s modified regimen) over 6 months. Established efficacy (complete/partial remission 70–80%); however, cyclophosphamide carries cumulative gonadotoxic, oncogenic (bladder cancer, lymphoma), and infectious risks. Now generally second-line after rituximab failure or in cases where rapid response is critical.
Cyclosporine/tacrolimus (calcineurin inhibitors): Reduce proteinuria effectively (70–80% response rate) but relapse is common after discontinuation (50–60%). Require careful monitoring of drug levels and renal function (nephrotoxicity). Now third-line or used in combination with rituximab for refractory cases.
Obinutuzumab: Type II anti-CD20 antibody (glycoengineered; more potent B-cell depletion than rituximab). GEMRITUX trial ongoing; used for rituximab-refractory MN in clinical practice at some centers.
Belimumab: Anti-BAFF/BLyS antibody (B-cell survival factor); being studied in combination with rituximab for maintenance of B-cell depletion and prevention of B-cell repopulation-driven relapse.
Anti-complement therapy: C5a receptor antagonists (avacopan) and other complement pathway inhibitors are under investigation, targeting the complement-mediated podocyte injury mechanism.
Adrenocorticotropic hormone (ACTH) gel: Synthetic ACTH (Acthar Gel, repository corticotropin) has been used in MN with modest evidence; mechanism may include melanocortin receptor–mediated podocyte protection beyond cortisol stimulation.
9. Complications
Venous thromboembolism (VTE): The dominant acute complication of MN. Renal vein thrombosis occurs in 25–50% of patients with heavy proteinuria and severe hypoalbuminemia. DVT and PE are also common. The mechanism involves urinary loss of natural anticoagulants (antithrombin III, proteins C and S) and hepatic upregulation of procoagulant factors. VTE may be asymptomatic or present as flank pain, hematuria, acute decline in GFR, or worsening proteinuria. Duplex ultrasound of renal veins is insensitive; CT or MR venography required to diagnose renal vein thrombosis.
Infections: Urinary losses of immunoglobulins and complement components create immune deficiency. Encapsulated organisms (pneumococcus, Haemophilus) and spontaneous bacterial peritonitis (if ascites is present) are particular risks. Pneumococcal and influenza vaccination is recommended for all nephrotic patients.
Cardiovascular disease: MN-associated nephrotic hyperlipidemia (elevated LDL and lipoprotein(a)) combined with CKD significantly increases atherosclerotic risk. Statin therapy is standard.
Progressive CKD: ~30% of primary MN patients progress to ESRD over 10–15 years. Risk factors include persistent heavy proteinuria, male sex, older age, hypertension, and advanced histological staging on biopsy.
Acute kidney injury: Can occur from bilateral renal vein thrombosis, severe volume depletion from aggressive diuresis, nephrotoxin exposure, or concurrent crescentic GN (rare). Bilateral thrombosis may present as sudden oliguria and worsening nephrotic syndrome.
Post-transplant recurrence: MN recurs in approximately 10–25% of transplanted kidneys. Anti-PLA2R or anti-THSD7A titers at the time of transplant predict recurrence risk; pretransplant anti-PLA2R reduction (with rituximab) is pursued in high-titer patients before transplantation.
10. Prognosis and the Rule of Thirds
The natural history of untreated primary MN over 10 years approximates the Rule of Thirds (described by Schieppati and Remuzzi):
- One-third achieve spontaneous complete remission (proteinuria <0.3 g/day, normal eGFR) — typically within 1–3 years of onset
- One-third achieve partial remission (proteinuria 0.3–3.5 g/day) with good long-term renal prognosis
- One-third have persistent nephrotic proteinuria progressing to ESRD within 10–15 years
Factors predicting spontaneous remission: female sex, younger age, lower initial proteinuria, undetectable or low anti-PLA2R titer. Factors predicting progression: male sex, older age, proteinuria >8 g/day, declining GFR, tubulointerstitial fibrosis on biopsy, and high anti-PLA2R titer.
With rituximab therapy, complete remission rates are substantially higher than with spontaneous remission alone: 40–60% complete remission in trials; partial remission in an additional 20–30%. Relapse after rituximab occurs in ~15–25% of responders, typically associated with B-cell repopulation and rising anti-PLA2R titers. Re-treatment with rituximab at the time of immunological relapse (titer rising, before proteinuria relapse) is an effective strategy.
Anti-PLA2R titer monitoring is the most useful tool for treatment guidance: immunological remission (titer undetectable) predicts protein remission with a lag of weeks to months. Patients who do not achieve immunological remission within 6–9 months of rituximab should be considered for re-treatment or alternative immunosuppression.
11. Research Papers (PubMed searches)
- Membranous nephropathy PLA2R antibody diagnosis
- Membranous nephropathy rituximab MENTOR trial
- Membranous glomerulonephritis complement C5b-9 podocyte injury
- Membranous nephropathy malignancy secondary causes
- Membranous nephropathy renal vein thrombosis anticoagulation
- THSD7A membranous nephropathy thyroid cancer
12. References
- Beck LH Jr, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361(1):11–21. PMID: 19571279. https://doi.org/10.1056/NEJMoa0810457
- Fervenza FC, et al. Rituximab or Cyclosporine in the Treatment of Membranous Nephropathy. N Engl J Med. 2019;381(1):36–46. PMID: 31269364. https://doi.org/10.1056/NEJMoa1814427
- Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021;100(4S):S1–S276. PMID: 34556256. https://doi.org/10.1016/j.kint.2021.05.021
- Ponticelli C, et al. A randomized trial of methylprednisolone and chlorambucil in idiopathic membranous nephropathy. N Engl J Med. 1989;320(1):8–13. PMID: 2642002. https://doi.org/10.1056/NEJM198901053200102
- Sethi S, et al. New concepts in the treatment of idiopathic membranous nephropathy. J Am Soc Nephrol. 2012;23(9):1417–1429. PMID: 22878962. https://doi.org/10.1681/ASN.2012020162
- Cattran DC, et al. Membranous nephropathy: quantifying remission duration on outcome. J Am Soc Nephrol. 2001;12(3):544–551. PMID: 11181800. https://doi.org/10.1681/ASN.V123544
- Tomas NM, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med. 2014;371(24):2277–2287. PMID: 25517707. https://doi.org/10.1056/NEJMoa1409354
- Remuzzi G, et al. Membranous nephropathy: risks versus benefits of therapeutic approaches. Lancet. 2001;358(9287):1133–1138. PMID: 11594254. https://doi.org/10.1016/S0140-6736(01)06260-0
- Schieppati A, Mosconi L, Perna A, et al. Prognosis of untreated patients with idiopathic membranous nephropathy. N Engl J Med. 1993;329(2):85–89. PMID: 8510707. https://doi.org/10.1056/NEJM199307083290203
- Hofstra JM, Fervenza FC, Wetzels JF. Treatment of idiopathic membranous nephropathy. Nat Rev Nephrol. 2013;9(8):443–458. PMID: 23799545. https://doi.org/10.1038/nrneph.2013.125
- Barbour SJ, et al. The MEST score provides earlier risk prediction in IgA nephropathy but requires renal biopsy. Kidney Int. 2016;89(1):167–175. PMID: 26475372. https://doi.org/10.1038/ki.2015.322
- Stanescu HC, et al. Risk HLA-DQA1 and PLA2R1 alleles in idiopathic membranous nephropathy. N Engl J Med. 2011;364(7):616–626. PMID: 21323541. https://doi.org/10.1056/NEJMoa1009742
Connections
- Nephrology & Hepatology
- Glomerulonephritis
- Nephrotic Syndrome
- IgA Nephropathy
- Focal Segmental Glomerulosclerosis
- Alport Syndrome
- Chronic Kidney Disease
- Rapidly Progressive GN
- Autoimmune Hepatitis
- Kidney Function Tests
- Urinalysis