Acromegaly
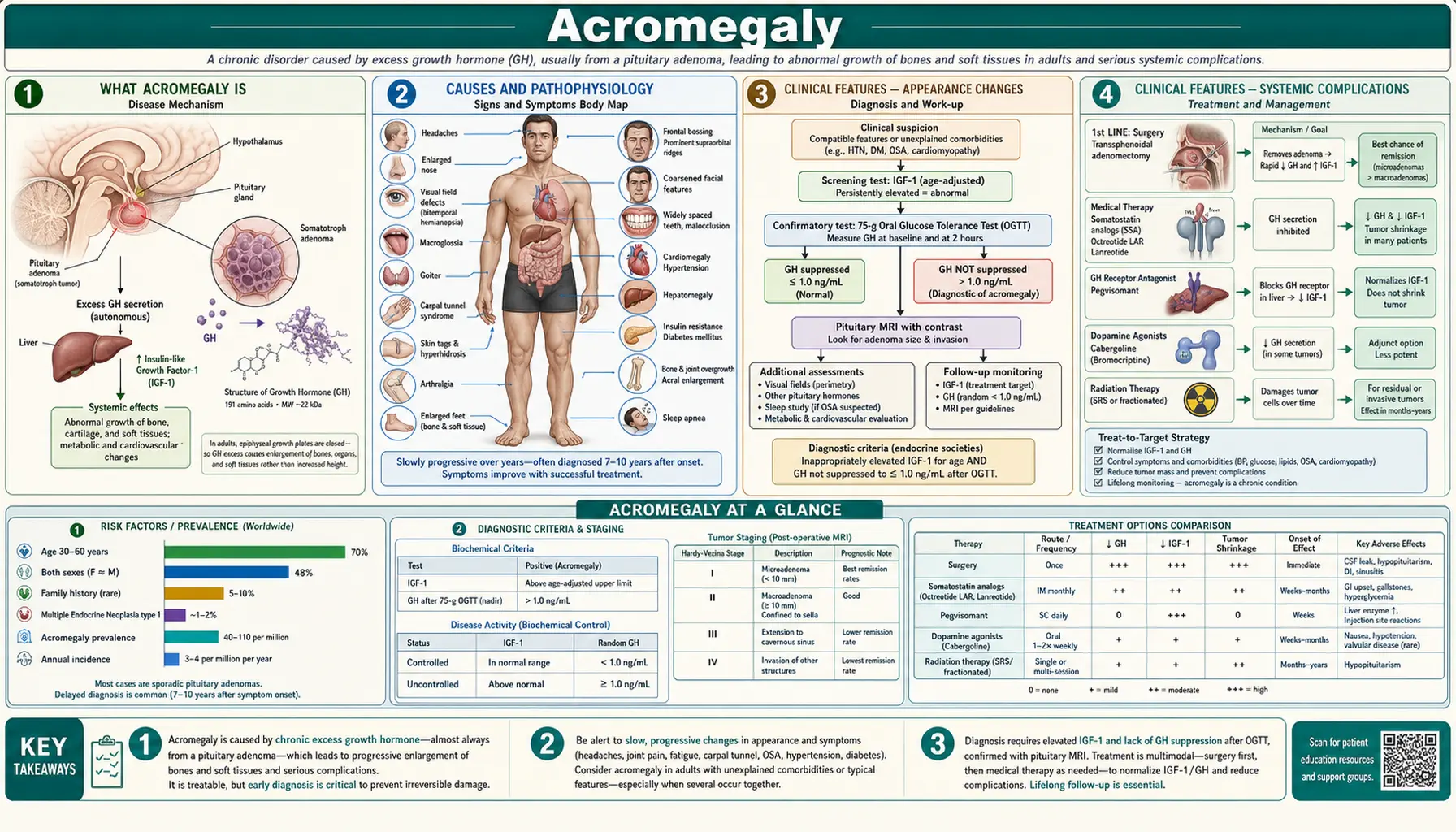
Acromegaly is a rare hormonal disorder in which the pituitary gland produces too much growth hormone (GH) in adults whose bones have already stopped lengthening. Because the growth plates are fused, the excess GH does not make you taller — instead it gradually thickens and enlarges bones, soft tissue, and internal organs over many years. Most people live with subtle changes for 7 to 10 years before a diagnosis is made, often noticing that rings no longer fit, shoe size has crept up, or facial features have coarsened. Left untreated, acromegaly raises the risk of heart disease, diabetes, sleep apnea, colorectal cancer, and early death. The good news is that modern surgery, medication, and radiation can control or cure the condition for the majority of patients.
Interactive Visualization Growth Hormone — lengthen a bone, then stop Pulse growth hormone from the pituitary to the liver, make IGF-1, and lengthen bone at the growth plates — then hit puberty, fuse the plates, and see why too much GH means gigantism or acromegaly. Launch →
Table of Contents
- What Acromegaly Is
- Causes and Pathophysiology
- Clinical Features — Appearance Changes
- Clinical Features — Systemic Complications
- Diagnosis — Biochemical Testing
- Diagnosis — Imaging
- Treatment — Surgery
- Treatment — Medical and Radiation Therapy
- Research Papers
- Connections
- Featured Videos
What Acromegaly Is
Growth hormone (GH) is produced by specialized cells called somatotrophs in the front part of the pituitary gland, a pea-sized structure at the base of the brain. GH does not act on bones and tissues directly. Instead, it travels to the liver and prompts it to release insulin-like growth factor 1 (IGF-1), which is the main driver of tissue growth. In healthy adults, the pituitary releases GH in short bursts — mainly during sleep — and IGF-1 levels stay within a narrow range. In acromegaly, this feedback system breaks down and GH pours into the bloodstream continuously, pushing IGF-1 far above normal.
When excess GH occurs before the growth plates close in adolescence, the result is gigantism — the skeleton grows abnormally tall. When it occurs after the growth plates have fused — as in all adults — the result is acromegaly. The word comes from the Greek akron (extremity) and megas (large), reflecting the most visible sign: enlargement of the hands, feet, and facial bones.
Acromegaly affects roughly 40–60 people per million worldwide. It is equally common in men and women, and most diagnoses are made between the ages of 40 and 50, though the disease typically began a decade earlier.
Causes and Pathophysiology
In approximately 95% of cases, acromegaly is caused by a benign, slow-growing tumor of the somatotroph cells in the pituitary gland — called a GH-secreting pituitary adenoma. These tumors are not cancerous and do not spread, but they grow autonomously, ignoring the body's normal signals to stop making GH. Adenomas smaller than 10 mm are called microadenomas; those 10 mm or larger are macroadenomas. At diagnosis, roughly 75% are macroadenomas, which explains why many patients already have mass-effect symptoms such as headache and vision changes by the time acromegaly is identified.
The remaining ~5% of cases arise from an excess of growth hormone–releasing hormone (GHRH), the brain chemical that normally tells the pituitary to secrete GH. GHRH-secreting tumors can occur in the hypothalamus, or — more commonly in this rare subset — in neuroendocrine neoplasms of the lung, pancreas, or gastrointestinal tract (carcinoid tumors). When a carcinoid tumor secretes GHRH ectopically, it chronically over-stimulates the pituitary somatotrophs, which can eventually cause diffuse somatotroph hyperplasia or a secondary adenoma.
Regardless of the trigger, the downstream mechanism is the same: continuously elevated GH drives the liver to overproduce IGF-1, and it is elevated IGF-1 — acting on receptors throughout the body — that causes nearly all of the clinical features of acromegaly. Understanding this GH → IGF-1 axis is essential for interpreting the diagnostic tests and choosing among treatments that target different steps in the pathway.
Clinical Features — Appearance Changes
The physical changes of acromegaly develop so slowly that patients and their families rarely notice them year to year. Looking at old photographs is often the most striking way to appreciate how much has changed over a decade. The changes fall into two main groups: acral changes (involving the extremities) and facial coarsening.
Acral (Extremity) Changes
- Hand enlargement: The hands become broad and fleshy. Rings that once fit comfortably no longer slide past the knuckle. A handshake feels noticeably "doughy" due to soft-tissue swelling.
- Foot enlargement: Shoe size increases by one or two sizes in adulthood — a change that has no other common explanation and is one of the most useful diagnostic clues in the history.
- Skin thickening: The skin becomes coarser, oilier, and thicker. Skin tags are common.
- Hyperhidrosis: Excessive sweating, often noticed on the palms, is reported by the majority of patients and tends to parallel disease activity — sweating improves when GH is controlled.
Facial Coarsening
- Prognathism and macrognathia: The lower jaw (mandible) grows forward and widens, creating an underbite and gaps between the teeth. Dental occlusion changes are often what prompt a dentist or orthodontist to first suspect the diagnosis.
- Macroglossia: The tongue enlarges, contributing to speech changes and, importantly, to airway narrowing during sleep.
- Frontal bossing: The brow ridges and forehead become more prominent as the frontal bones thicken.
- Nasal broadening: The nose widens and the nasal bridge thickens.
- Supraorbital ridge prominence: The eyebrow ridges protrude, giving the face a heavier, more angular appearance.
Because these changes occur over years, patients are often shocked when they compare a current photograph with one from 5–10 years earlier. The transformation is real but gradual, which is why the average time from symptom onset to diagnosis is 7–10 years.
Clinical Features — Systemic Complications
Acromegaly is much more than a cosmetic condition. Elevated IGF-1 acts on nearly every organ system, and it is the systemic complications — not the appearance changes — that drive the increased mortality seen in untreated disease. The standardized mortality ratio in uncontrolled acromegaly is approximately 1.5 to 3 times that of the general population.
Cardiovascular Disease
The heart is one of the most important targets of excess IGF-1. Cardiomegaly (enlarged heart) and left ventricular hypertrophy (LVH) occur in the majority of patients with long-standing disease. Valvular abnormalities — particularly aortic and mitral regurgitation — are more common than in the general population. Hypertension affects roughly 35–40% of patients. Biventricular dysfunction and heart failure are serious consequences of late-stage, untreated acromegaly. Cardiovascular disease is the leading cause of death in acromegaly.
Sleep Apnea
Sleep apnea — both obstructive (due to macroglossia and pharyngeal soft-tissue enlargement) and central (due to hypothalamic effects of GH) — occurs in roughly 80% of patients. This is strikingly high and means that acromegaly should be considered whenever a patient presents with severe, treatment-resistant sleep apnea alongside any physical features of GH excess.
Glucose Intolerance and Diabetes
GH is a counter-regulatory hormone that opposes insulin. Sustained GH excess induces hepatic and peripheral insulin resistance. Diabetes mellitus or impaired glucose tolerance develops in 25–40% of patients. Hyperglycemia often improves substantially after effective treatment of acromegaly, though it may not fully normalize if beta-cell damage has occurred.
Arthropathy
Joint pain and arthritis are among the most disabling symptoms for many patients. Excess GH stimulates cartilage and periarticular soft-tissue overgrowth, leading to a distinctive acromegalic arthropathy affecting the knees, hips, spine, and shoulders. Unlike standard osteoarthritis, the joint space may initially appear widened on X-ray (due to cartilage hypertrophy) before eventually narrowing as the cartilage breaks down. Arthropathy often persists or even progresses after biochemical control is achieved, making it one of the most refractory complications.
Carpal Tunnel Syndrome
Carpal tunnel syndrome occurs in approximately 30% of patients, caused by soft-tissue swelling within the carpal tunnel that compresses the median nerve. Numbness, tingling, and weakness in the hand are the classic symptoms. It often improves with treatment of the underlying GH excess.
Colonic Polyps and Colorectal Cancer Risk
IGF-1 promotes cell proliferation in the colonic mucosa. Patients with acromegaly have a 2 to 3 times higher risk of colorectal cancer compared with the general population and a higher prevalence of colonic polyps (both adenomatous and hyperplastic). Current guidelines recommend colonoscopy at diagnosis and regular follow-up surveillance colonoscopies thereafter.
Visceromegaly
The thyroid, liver, kidneys, spleen, and salivary glands can all enlarge under the influence of excess IGF-1. Thyroid goiter is particularly common and may cause compressive symptoms in the neck.
Headache and Visual Field Loss
The growing pituitary tumor itself causes two important symptoms. Headache — often retro-orbital or bifrontal — is reported by the majority of patients with macroadenomas. When the tumor grows upward and compresses the optic chiasm (the crossing point of the optic nerves), patients develop bitemporal hemianopia: loss of the outer (temporal) visual fields in both eyes. Patients sometimes describe bumping into door frames or not seeing people approaching from the side. This is a medical urgency because it can progress to permanent blindness if the tumor is not decompressed promptly.
Diagnosis — Biochemical Testing
Diagnosing acromegaly requires biochemical confirmation of GH excess. Clinical features alone are insufficient because of the broad differential for facial and acral changes, and because the appearance evolves so slowly that both patient and doctor may not recognize it.
Why Random GH Is Unreliable
GH is secreted in pulses, primarily during sleep. In healthy people, GH levels between pulses can be nearly undetectable (<0.1 ng/mL). Because acromegaly causes chronically elevated rather than uniformly elevated GH, a single random measurement can be normal even in active disease. Conversely, stress, exercise, or acute illness can transiently raise GH in healthy people, leading to false positives. For these reasons, random serum GH is not a useful screening or diagnostic test for acromegaly.
IGF-1: The Preferred Screening Test
Serum IGF-1 is the best single test for acromegaly. Because IGF-1 is produced continuously (not in pulses), it provides a stable, integrated measure of GH secretion over the preceding 24 hours. A single morning fasting blood draw is all that is needed. The result must be interpreted against age- and sex-matched reference ranges, because IGF-1 is naturally highest in adolescence and declines with age. An elevated IGF-1 in an adult, after excluding physiological causes (pregnancy, puberty, liver disease, malnutrition — all of which alter IGF-1 independently), is highly suggestive of acromegaly.
Oral Glucose Tolerance Test (OGTT) — The Confirmatory Test
The definitive biochemical test is the OGTT-GH suppression test. The patient drinks 75 g of glucose, and GH is measured at 0, 30, 60, 90, and 120 minutes. In healthy people, the glucose load suppresses GH to below 1 ng/mL (some guidelines use <0.4 ng/mL with modern ultrasensitive assays) within 60–120 minutes. In acromegaly, GH fails to suppress — it remains elevated throughout, and in many cases paradoxically rises. This failure of normal GH suppression during an OGTT is the diagnostic hallmark of acromegaly.
The OGTT is the gold standard because it directly tests the pituitary's ability to respond to the normal physiological brake on GH secretion. It is particularly important when IGF-1 is borderline or when there are other reasons that might affect IGF-1 levels.
Diagnosis — Imaging
Once biochemical testing confirms GH excess, the next step is to locate the source with imaging. In the vast majority of cases, the culprit is a pituitary adenoma, and MRI of the pituitary with gadolinium contrast is the imaging modality of choice.
Pituitary MRI with Gadolinium
Gadolinium-enhanced MRI provides excellent soft-tissue contrast for the pituitary gland and surrounding structures. The images are reviewed in coronal and sagittal planes with thin (2–3 mm) cuts through the sella turcica. Most GH-secreting adenomas enhance less brightly than the normal pituitary gland after gadolinium injection, making them visible as a relatively hypointense area within the gland. Dynamic gadolinium sequences (multiple images taken rapidly after contrast injection) can improve detection of very small tumors.
Microadenoma vs Macroadenoma
Microadenomas (<10 mm) are smaller and typically confined within the sella turcica. They have higher surgical cure rates (up to 80%) because a skilled neurosurgeon can usually resect the entire tumor without damaging the normal pituitary. Macroadenomas (≥10 mm) are more challenging. They may extend upward into the suprasellar space (where they can compress the optic chiasm), laterally into the cavernous sinuses (which house major blood vessels and cranial nerves), or inferiorly into the sphenoid sinus. Cavernous sinus invasion, which can be assessed with MRI using criteria such as the Knosp grade, is the most important predictor of surgical failure because the cavernous sinus cannot be safely entered without high risk of major hemorrhage or cranial nerve injury.
When No Pituitary Tumor Is Found
If biochemical testing strongly suggests acromegaly but MRI shows a normal pituitary, ectopic GHRH secretion from a lung, pancreatic, or gastrointestinal neuroendocrine tumor should be considered. In this situation, serum GHRH levels should be measured, and imaging of the chest, abdomen, and pelvis (CT or octreotide scintigraphy/DOTATATE PET) is performed to locate the primary tumor.
Treatment — Surgery
For most patients with acromegaly caused by a pituitary adenoma, surgery is the recommended first-line treatment. When successful, surgery provides an immediate, durable cure by removing the source of GH excess.
Transsphenoidal Surgery
The standard surgical approach is transsphenoidal surgery, which reaches the pituitary gland through the nose and sphenoid sinus without any external incision or brain retraction. Modern endoscopic transsphenoidal techniques use a high-definition camera and angled instruments to give the neurosurgeon a wide, illuminated view of the tumor and surrounding structures. The procedure typically takes 2–4 hours, and most patients are discharged within 2–3 days.
Cure Rates
Surgical cure rates depend strongly on tumor size and extent. For microadenomas, cure — defined as a normalized IGF-1 and GH suppression to <1 ng/mL on OGTT — is achieved in approximately 75–85% of cases by experienced pituitary surgeons. For macroadenomas, cure rates fall to roughly 40–55%, largely because these tumors are more likely to invade the cavernous sinus where complete resection is impossible. Surgical outcomes are highly operator-dependent: case volume matters, and patients with acromegaly should ideally be referred to centers that perform at least 50 pituitary surgeries per year.
Residual and Recurrent Disease
Patients who are not cured by surgery — meaning their IGF-1 and GH levels remain elevated postoperatively — have residual disease and require adjuvant treatment. Surgical remission is assessed biochemically at 12 weeks after the operation (earlier measurements may be misleading due to transient changes in GH dynamics). True recurrence after an initial biochemical cure is uncommon but does occur, particularly in the first 5 years; lifelong follow-up is therefore recommended even for patients who achieve remission.
Surgical Risks
In experienced hands, transsphenoidal surgery is safe. The most important risks include: hypopituitarism (damage to normal pituitary tissue causing deficiencies of other hormones such as cortisol, thyroid hormone, or sex hormones), diabetes insipidus (damage to the posterior pituitary or stalk causing loss of the anti-diuretic hormone), cerebrospinal fluid leak (which requires repair to prevent meningitis), and, very rarely, major vascular injury. Rates of serious complications are below 2% at high-volume centers.
Treatment — Medical and Radiation Therapy
Medical therapy is used when surgery fails to achieve biochemical control, when a patient has contraindications to surgery, or when pre-operative tumor shrinkage is desired. Three pharmacological classes are available, each targeting a different point in the GH/IGF-1 pathway.
Somatostatin Analogues (SSAs)
Somatostatin is the brain hormone that normally inhibits GH secretion. Long-acting somatostatin analogues — octreotide LAR (given as a monthly intramuscular injection) and lanreotide autogel (a monthly deep subcutaneous injection) — mimic this natural brake. They normalize IGF-1 in approximately 55–65% of patients and shrink the tumor in about 50%. SSAs are typically the first pharmacological choice because they simultaneously target both GH secretion and tumor volume. Side effects include gallstones (the most common long-term complication, occurring in 20–30% of patients on chronic SSA therapy), nausea, diarrhea, and flatulence. A newer oral somatostatin analogue (octreotide capsules) was approved in 2020 for patients who have already responded to injectable SSAs.
Pegvisomant — GH Receptor Antagonist
Pegvisomant has a completely different mechanism: instead of suppressing GH secretion, it blocks the GH receptor in peripheral tissues, preventing GH from stimulating IGF-1 production. It is given as a daily subcutaneous injection. Pegvisomant normalizes IGF-1 in over 90% of patients and is the most effective drug available for lowering IGF-1 — but it does not reduce GH levels (which may actually rise slightly) or shrink the tumor. It is primarily used in patients whose IGF-1 is not controlled by SSAs, often in combination with them. Liver enzyme elevations occur in a small proportion of patients and require monitoring.
Cabergoline
Cabergoline is a dopamine agonist used primarily for prolactin-secreting tumors (prolactinomas). It also suppresses GH in some patients with acromegaly — particularly those whose tumor co-secretes prolactin — and has the convenience of twice-weekly oral administration. It is best suited for patients with mild IGF-1 elevations (up to ~1.5–2 times the upper limit of normal) and is less potent than SSAs for GH control, but substantially cheaper and easier to take.
Stereotactic Radiosurgery
Radiation therapy is reserved for patients who have not been cured by surgery and who cannot achieve biochemical control with medication alone, or who refuse or cannot tolerate surgery. Stereotactic radiosurgery (Gamma Knife, CyberKnife, or linear accelerator-based radiosurgery) delivers a precisely focused, high dose of radiation to the residual adenoma in a single session. Biochemical remission rates of 40–60% are reported, but the critical limitation is time: it typically takes 5–15 years for GH and IGF-1 to normalize after radiosurgery, during which continued medical therapy is required. The major risk is hypopituitarism, which develops in 20–50% of patients over 10 years. Conventional fractionated radiotherapy (delivered in small daily doses over 5–6 weeks) is an alternative when the tumor is too large or too close to the optic chiasm for single-session radiosurgery, but it carries even higher rates of hypopituitarism and cognitive effects.
Combination Approaches and Monitoring
Many patients with macroadenomas require a combination of surgery, SSA, and possibly radiosurgery to achieve long-term biochemical control. Treatment targets are IGF-1 within the age-matched normal range and GH <1 ng/mL (or <0.4 ng/mL by ultrasensitive assay). Patients also need long-term monitoring for complications — annual echocardiography, colonoscopy at diagnosis with repeat every 5–10 years, fasting glucose and HbA1c, and thyroid function — because many systemic complications persist even after GH normalization.
Research Papers
- Melmed S (2019). Acromegaly pathogenesis and treatment. J Clin Invest, 129(1):1229 — Search PubMed. doi:10.1172/JCI134360
- Katznelson L, et al (2014). Acromegaly: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 99(11):3933–3951. PMID 25356808. doi:10.1210/jc.2014-2700
- Colao A, Ferone D, Marzullo P, Lombardi G (2004). Systemic complications of acromegaly. Endocr Rev, 25(1):102–152 — Search PubMed. doi:10.1210/er.2002-0015
- Holdaway IM, Rajasoorya RC, Gamble GD (2004). Factors influencing mortality in acromegaly. J Clin Endocrinol Metab, 89(2):667–674 — Search PubMed. doi:10.1210/jc.2003-031199
- Freda PU, et al (2011). Pituitary Incidentaloma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 96(4):894–904. PMID 21474687. doi:10.1210/jc.2010-1048
- Giustina A, et al (2014). Expert consensus document: A consensus on the diagnosis and treatment of acromegaly comorbidities. Pituitary, 17(5):491–503 — Search PubMed. doi:10.1007/s11102-013-0545-7
- Sherlock M, Ayuk J, Tomlinson JW, et al (2010). Mortality in patients with pituitary disease. Endocr Rev, 31(3):301–342 — Search PubMed. doi:10.1210/er.2009-0033
- van der Lely AJ, et al (2001). Biochemical assessment of disease activity and therapeutic outcome in acromegaly. Eur J Endocrinol, 144(3):223–227 — Search PubMed. doi:10.1530/eje.0.1440223
- Melmed S, et al (2018). Guidelines for acromegaly management: an update. J Clin Endocrinol Metab, 93(4):1932 — Search PubMed. doi:10.1210/jc.2017-01703
- Chanson P, Salenave S (2008). Acromegaly. Orphanet J Rare Dis, 3:17 — Search PubMed. doi:10.1186/1750-1172-3-17
- Trainer PJ, et al (2000). Treatment of acromegaly with the growth hormone–receptor antagonist pegvisomant. N Engl J Med, 342(16):1171–1177. PMID 10770982. doi:10.1056/NEJM200004203421604
- Nomikos P, Buchfelder M, Fahlbusch R (2005). The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical cure. Eur J Endocrinol, 152(3):379–387. PMID 15757854. doi:10.1530/eje.1.01863
Connections
- Endocrinology
- Growth Hormone: How You Grow (and Why It Stops) — interactive animation
- Cushing's Syndrome
- Prolactinoma
- Hypothyroidism
- Type 2 Diabetes
- Pheochromocytoma
- Neurology Conditions
- Lab Tests