NAD+ & NMN for Longevity and Sirtuin Activation
Of all the candidate "longevity supplements" on the market, NAD+ precursors are the only ones tied to a coherent and rigorously tested biological theory of aging. The David Sinclair lab at Harvard Medical School established the connection between NAD+ availability and the sirtuin family of enzymes, and the resulting model — declining NAD+ silences sirtuins, which in turn allows epigenetic decay and mitochondrial dysfunction to accumulate — remains the dominant scientific framework for thinking about NAD+ supplementation in 2026. This deep-dive walks through the seven mammalian sirtuins, the SIRT1/SIRT3/SIRT6 axis that does most of the heavy longevity lifting, the Sinclair Information Theory of Aging, the Brenner vs Sinclair NR-vs-NMN debate, and an honest assessment of what is and is not proven in humans.
Table of Contents
- The NAD+ Decline = Aging Hypothesis
- The Sirtuin Family (SIRT1–SIRT7)
- SIRT1 — Caloric Restriction Mimicry and Autophagy
- SIRT3 — Mitochondrial Sentinel
- SIRT6 — Telomeres, DNA Repair, and Lifespan
- The David Sinclair Lab Program
- The Information Theory of Aging
- The Brenner vs Sinclair NR-vs-NMN Debate
- What Is and Is Not Proven in Humans
- A Practical Sirtuin-Activating Stack
- Cautions
- Key Research Papers
- Connections
The NAD+ Decline = Aging Hypothesis
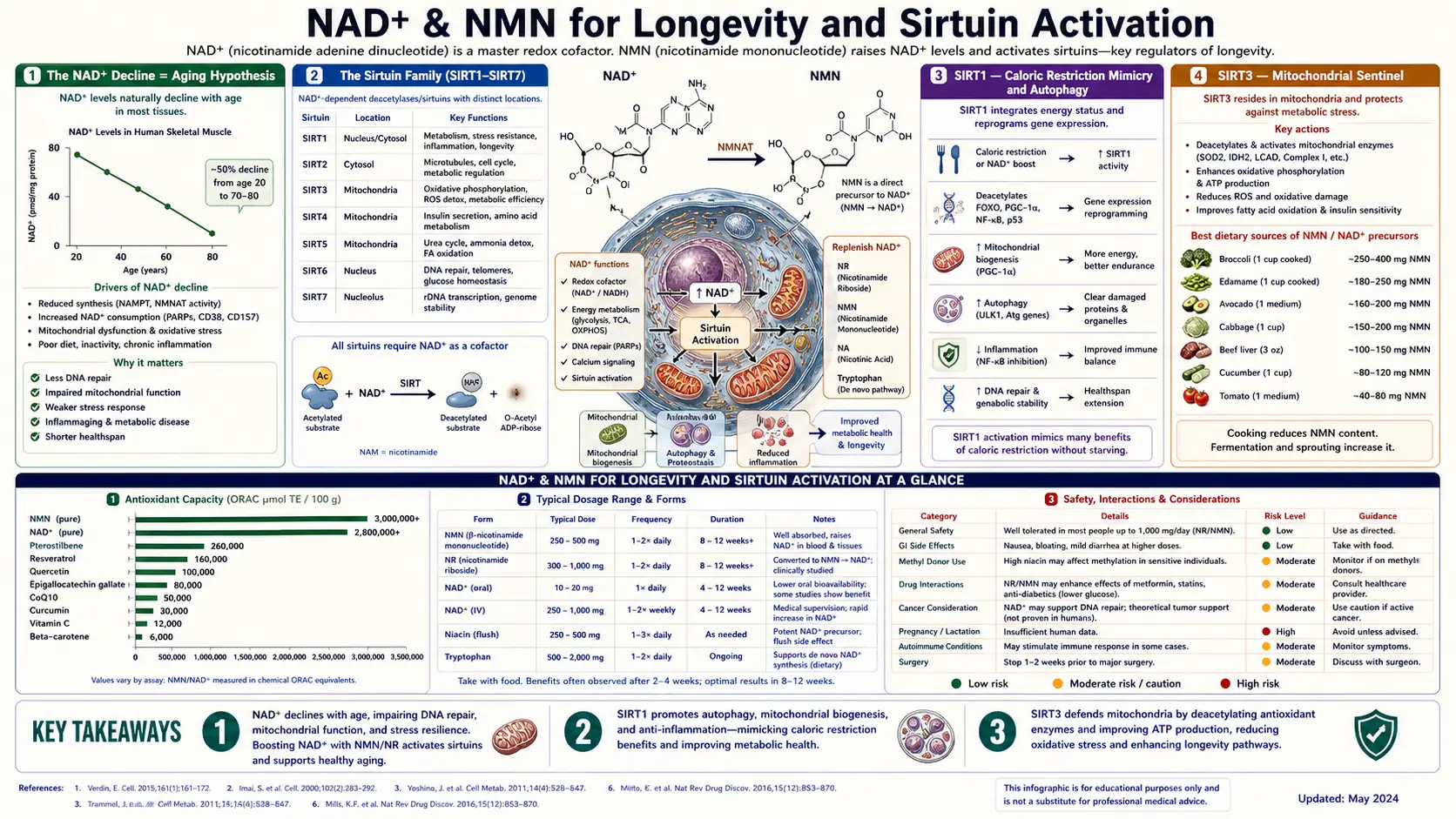
The central premise of NAD+-centric longevity science is straightforward: NAD+ concentrations in human tissues decline by roughly 50% between young adulthood and middle age, and by even more in older age. This decline is documented in skin, blood, liver, skeletal muscle, and brain, and it correlates with multiple hallmarks of aging including mitochondrial dysfunction, genomic instability, cellular senescence, chronic inflammation, and impaired stem-cell function.
The hypothesis is that this NAD+ decline is not just a marker of aging but a cause of it. The reasoning runs as follows:
- Sirtuins, PARPs, and CD38 all consume NAD+ as a stoichiometric substrate.
- As DNA damage accumulates with age, PARPs consume more NAD+ to flag and repair breaks.
- As inflammaging develops, CD38 expression on inflammatory immune cells rises, consuming more NAD+.
- Meanwhile, the rate-limiting NAD+ salvage enzyme NAMPT loses activity with age, reducing supply.
- The net result is a falling pool of NAD+ available to the sirtuins.
- Sirtuins, starved of substrate, can no longer maintain their normal epigenetic, metabolic, and DNA-repair functions.
- Cellular function degrades, contributing to the visible phenotype of aging.
If the chain is correct, restoring NAD+ to youthful levels should reactivate sirtuin function and slow or partially reverse aspects of biological aging. This is the scientific frame within which all NMN and NR supplementation should be understood — not as a vitamin replacement, but as a substrate replenishment for a class of enzymes whose substrate has become limiting.
The hypothesis is well supported by mouse data and by the documented age-related decline in NAD+ itself, but the final step — that boosting NAD+ in humans produces measurable extension of healthspan or lifespan — remains an active research question. Short human trials show reliable NAD+ elevation; longer trials examining functional aging outcomes are underway in 2026.
The Sirtuin Family (SIRT1–SIRT7)
Mammals have seven sirtuins, named SIRT1 through SIRT7. All seven require NAD+ as a stoichiometric cofactor — one NAD+ molecule is consumed for each deacetylation reaction. They differ in subcellular location and substrate preference, which determines their specific roles in cellular biology.
| Sirtuin | Location | Primary Activities |
|---|---|---|
| SIRT1 | Nucleus, cytoplasm | Deacetylates p53, FOXO, PGC-1α, NF-kB; promotes autophagy; mediates caloric restriction |
| SIRT2 | Cytoplasm | Tubulin deacetylase; cell-cycle control; oligodendrocyte function |
| SIRT3 | Mitochondrial matrix | Master mitochondrial deacetylase; regulates electron transport, antioxidant defenses, fatty acid oxidation |
| SIRT4 | Mitochondrial matrix | ADP-ribosyltransferase rather than deacetylase; regulates glutamate dehydrogenase |
| SIRT5 | Mitochondrial matrix | Demalonylates, desuccinylates, deglutarylates rather than classic deacetylation |
| SIRT6 | Nucleus, chromatin | DNA double-strand break repair, telomere maintenance, glucose metabolism regulation |
| SIRT7 | Nucleolus | Ribosomal DNA stability, transcription regulation |
For longevity purposes, the three that matter most are SIRT1 (the master metabolic sensor), SIRT3 (the mitochondrial protector), and SIRT6 (the chromatin guardian). The next three sections examine each in detail.
SIRT1 — Caloric Restriction Mimicry and Autophagy
SIRT1 is the human homolog of yeast Sir2, the original sirtuin discovered by Leonard Guarente at MIT in 1999. Its activity was the first molecular explanation for why caloric restriction extends lifespan in yeast, worms, flies, and mice: caloric restriction raises the cellular NAD+/NADH ratio, activating SIRT1, which then deacetylates dozens of downstream targets to coordinate the longevity-associated metabolic shift.
Key SIRT1 substrates and their downstream effects:
- p53 — SIRT1 deacetylates p53 at lysine 382, reducing its pro-apoptotic activity and shifting cells toward survival rather than death under stress. This is part of how caloric restriction protects against age-related cell loss.
- FOXO transcription factors — SIRT1 deacetylation of FOXO1, FOXO3, and FOXO4 promotes their stress-resistance and antioxidant gene programs, including superoxide dismutase 2 (SOD2) and catalase. This is a major reason SIRT1 activation enhances cellular resilience.
- PGC-1α — SIRT1 deacetylates and activates PGC-1α, the master regulator of mitochondrial biogenesis. The result is more mitochondria per cell, better-organized respiratory chains, and improved oxidative metabolism — the same phenotype induced by exercise.
- NF-kB — SIRT1 deacetylates the p65 subunit of NF-kB, reducing inflammatory gene transcription. This is the molecular basis for SIRT1's anti-inflammaging effects.
- ATG proteins and LC3 — SIRT1 promotes the assembly of the autophagy machinery, enabling cells to clear damaged proteins and organelles.
The implication for NAD+ supplementation is direct: SIRT1 activity is rate-limited by NAD+ availability under normal conditions, so raising the NAD+ pool with NMN or NR should mechanistically enhance SIRT1 function and produce caloric-restriction-like effects without the caloric restriction itself. This is the basis for the framing of NMN as a "caloric restriction mimetic."
SIRT3 — Mitochondrial Sentinel
SIRT3 lives in the mitochondrial matrix and deacetylates a remarkably large fraction of all mitochondrial proteins — estimates suggest it regulates over 60% of the mitochondrial proteome. Its substrates include:
- Long-chain acyl-CoA dehydrogenase (LCAD) — SIRT3 deacetylates LCAD, enabling efficient fatty acid oxidation. SIRT3 knockout mice cannot oxidize long-chain fatty acids properly and develop fatty liver under fasting.
- Manganese superoxide dismutase (MnSOD/SOD2) — SIRT3 deacetylates SOD2, activating the principal mitochondrial antioxidant defense. Reduced SIRT3 activity (as in aged tissues) leaves mitochondrial superoxide unchecked.
- Complex I and Complex II subunits — SIRT3 deacetylates respiratory chain subunits, supporting normal electron flow and ATP production.
- Isocitrate dehydrogenase 2 (IDH2) — SIRT3 deacetylation of IDH2 supports NADPH regeneration, which is required for glutathione recycling.
The Sinclair lab and others have shown that CD38, the NAD+ consumer that rises with age, drives the age-related decline in SIRT3 activity through NAD+ depletion. Reduced SIRT3 activity then directly causes the mitochondrial dysfunction observed in aged tissues — reduced ATP production, increased ROS leak, and impaired fatty acid oxidation. NAD+ repletion targets this pathway by giving SIRT3 the substrate it needs.
SIRT6 — Telomeres, DNA Repair, and Lifespan
SIRT6 is perhaps the most directly "longevity-relevant" sirtuin. Mice engineered to overexpress SIRT6 live measurably longer than wild-type controls, and SIRT6 knockout mice die prematurely with a phenotype resembling accelerated aging.
SIRT6 has three principal functions:
- Telomere chromatin maintenance — SIRT6 deacetylates histone H3 at lysine 9 (H3K9ac) and lysine 56 (H3K56ac) at telomeric chromatin, supporting the compact heterochromatin structure that protects chromosome ends. Without SIRT6, telomeric chromatin destabilizes and chromosomal ends become dysfunctional.
- DNA double-strand break repair via homologous recombination — SIRT6 is recruited to DNA breaks and facilitates the homologous recombination pathway, which is the higher-fidelity repair mechanism. Reduced SIRT6 activity shifts repair toward the more error-prone non-homologous end joining pathway, producing accumulating mutations.
- Glucose metabolism regulation — SIRT6 represses glycolytic genes via deacetylation of H3K9 at glycolysis-related promoters, biasing metabolism toward oxidative phosphorylation. Loss of SIRT6 produces a Warburg-like shift toward glycolysis, which is metabolically associated with both cancer and aging.
SIRT6 activity, like SIRT1 and SIRT3 activity, is NAD+-dependent. Restoring NAD+ levels in aged tissues with NMN or NR is expected to reactivate SIRT6 alongside the other sirtuins, supporting all three functions. The telomere and DNA repair effects in particular are the mechanistic basis for the "NAD+ extends healthspan" framing in mouse studies.
The David Sinclair Lab Program
David Sinclair is the tenured Professor of Genetics at Harvard Medical School whose lab has done more than any other to establish the scientific case for NAD+-based interventions. His research arc moves through three phases:
Phase 1: The Sir2 / SIRT1 discovery (1995–2003)
Sinclair completed his Ph.D. in Molecular Genetics at the University of New South Wales in 1995, then joined Leonard Guarente's lab at MIT as a postdoctoral fellow. There he co-discovered a cause of yeast aging tied to genomic instability at the ribosomal DNA locus, and identified Sir2 (the yeast sirtuin) as a key regulator of that process. He founded his own lab at Harvard Medical School in 1999 and extended the Sir2 work into mammalian SIRT1.
Phase 2: Resveratrol and the STAC search (2003–2013)
Sinclair's 2003 demonstration that resveratrol activates yeast Sir2 and extends yeast lifespan launched a decade-long search for sirtuin-activating compounds (STACs). The resveratrol findings were controversial — some labs failed to replicate certain mechanistic claims — but the field has since established that synthetic STACs (such as SRT2104) and natural compounds (such as pterostilbene) can activate SIRT1 in a substrate-dependent manner. Sinclair co-founded Sirtris Pharmaceuticals around this work; Sirtris was acquired by GlaxoSmithKline in 2008.
Phase 3: NAD+ precursors and epigenetic reprogramming (2013–present)
Recognizing that STACs only work when their substrate (NAD+) is available, Sinclair's lab turned to NAD+ precursors. The 2013 paper from his group (Gomes et al., Cell) showed that NMN administration to aged mice restored mitochondrial function in skeletal muscle within one week. Mills et al. 2016 (Imai lab at Washington University, closely tied to Sinclair) showed that long-term NMN supplementation mitigated age-related physiological decline in mice across multiple organ systems. Sinclair's lab has more recently focused on cellular reprogramming as a more direct intervention against the epigenetic noise that he believes drives aging, while continuing to publish on NAD+ biology.
Sinclair has consistently advocated for NMN over NR in his public commentary, in part because his own lab's data are NMN-centric. He has personally reported taking 1 g of NMN daily along with resveratrol, metformin, and other compounds. His advocacy has been instrumental in driving consumer interest in NMN supplementation — for better or worse, depending on whose interpretation one accepts (see the Brenner debate below).
The Information Theory of Aging
In his 2019 book Lifespan and a series of subsequent papers, Sinclair proposed a unifying framework he calls the Information Theory of Aging. The core idea: aging is fundamentally a loss of epigenetic information, not genetic information. Cells lose the ability to read their own DNA correctly because the epigenetic marks that tell each cell which genes to express become progressively scrambled over time.
The mechanism proposed is that DNA double-strand breaks — which occur thousands of times per cell per day from normal metabolism, radiation, and chemical stress — recruit sirtuins (particularly SIRT1) away from their normal epigenetic maintenance duties to participate in emergency repair. Each repair episode subtly perturbs the local chromatin state and leaves a small amount of epigenetic noise behind. Over decades, this accumulating noise progressively degrades cellular identity, leading to functional decline.
The implications for NAD+ supplementation under this model:
- NAD+ supplementation should reduce the "tax" that DNA damage imposes on sirtuin availability, allowing sirtuins to maintain their epigenetic maintenance functions more effectively.
- NAD+ supplementation supports SIRT6, which directs DNA repair toward the higher-fidelity homologous recombination pathway, reducing the rate of epigenetic perturbation per repair event.
- Cellular reprogramming with the Yamanaka transcription factors (OSK, omitting cMyc) may directly reverse epigenetic noise, restoring a more youthful epigenetic state — this is now the main focus of Sinclair's laboratory.
The theory is not universally accepted; competing frameworks emphasize mitochondrial decline, telomere attrition, senescent cell accumulation, or stem cell exhaustion as the primary drivers. But the Information Theory has been productive scientifically and has provided a coherent rationale for the otherwise disparate sirtuin/NAD+/epigenetic-reprogramming research programs at the Sinclair lab.
The Brenner vs Sinclair NR-vs-NMN Debate
Charles Brenner, the biochemist who discovered the NR pathway in 2004 and who is the chief scientific advisor at ChromaDex (the company that markets NR as Niagen), has been the most prominent scientific critic of the NMN-over-NR framing. The Brenner critique has several components, of which the most consequential are:
- Bioavailability uncertainty — Brenner argues that NMN, being a charged phosphorylated molecule, cannot enter cells directly and must first be dephosphorylated extracellularly to nicotinamide riboside, then transported in via NR kinases, then re-phosphorylated to NMN inside the cell. If true, this means oral NMN and oral NR end up doing the same thing biochemically — with NR being more direct and therefore more bioavailable per mg administered. Sinclair's lab counters that the Slc12a8 NMN transporter in intestinal cells allows direct NMN uptake, although this transporter's broader role outside the intestine remains contested.
- Methylation cofactor depletion — All NAD+ precursors that pass through nicotinamide as a metabolic intermediate (which includes both NR and NMN) increase the demand for methylation. The body methylates excess nicotinamide (via NNMT) to N1-methylnicotinamide before excretion. High-dose, chronic NMN or NR supplementation can therefore deplete the methyl donor pool, potentially affecting DNA methylation, neurotransmitter synthesis, and homocysteine. This concern argues for co-supplementing with a methyl donor such as TMG (trimethylglycine, 500–1,000 mg/day) when taking high-dose NMN or NR.
- Trial evidence asymmetry — As of 2026, more human RCTs have used NR than NMN, and NR has a longer regulatory and safety history (it was on the US market continuously since 2013 as Niagen). NMN's human evidence base is growing rapidly but is still smaller.
- Commercial conflicts — Both sides have commercial interests: Brenner is tied to ChromaDex (NR); Sinclair co-founded Metro International Biotech (pharmaceutical NMN as MIB-626). Readers should weigh the published data on its scientific merits rather than the public personalities involved.
The most honest synthesis as of 2026 is that both NR and NMN reliably raise blood NAD+ levels in humans, that the size of the increase per mg dose is broadly similar, and that the choice between them is largely a matter of cost, availability, and personal preference. A 2026 head-to-head clinical trial reported that both NR and NMN doubled circulating NAD+ over 14 days while plain nicotinamide (NAM) did not, suggesting the two precursors are functionally interchangeable for the NAD+-raising endpoint.
What Is and Is Not Proven in Humans
Honest accounting of the 2026 human evidence base:
Well established
- NMN and NR reliably raise blood NAD+ levels and NAD+ metabolites in a dose-dependent manner.
- Both precursors are safe and well tolerated at doses up to 1,000–2,000 mg/day for up to 24 weeks.
- NMN supplementation increases muscle insulin sensitivity in prediabetic women (Yoshino Science 2021).
- NR modestly reduces systolic blood pressure and aortic stiffness in healthy middle-aged adults (Martens 2018).
Promising but not yet proven
- Improvements in physical performance (Uthever multicenter trial, NMN runner trial).
- Improvements in subjective health scores and walking endurance.
- Cardiovascular benefits beyond blood pressure (LDL, inflammation, endothelial function).
Not yet demonstrated in humans
- Extension of lifespan (no human trial has run long enough to measure this; mouse data is encouraging).
- Reversal of established cognitive decline.
- Reversal of established frailty or sarcopenia.
- Improved fertility in older women (mouse data is striking, but human trials are not yet completed).
- Slowing of biological-age clocks at the population level.
The conservative reading: NMN and NR are safe and reliably do what they are supposed to do biochemically (raise NAD+). Whether this translates to clinically meaningful longevity benefit in humans is plausible but not yet proven, and individual responses will likely vary substantially based on baseline NAD+ status, age, and the burden of CD38-driven inflammaging.
A Practical Sirtuin-Activating Stack
For those who have weighed the evidence and chosen to supplement, the most-discussed sirtuin-supporting stack in 2026 includes:
- NMN or NR 250–1,000 mg/day — taken in the morning on an empty stomach, aligned with the natural circadian peak of NAD+ metabolism
- TMG (trimethylglycine) 500–1,000 mg/day — methyl donor to offset the methylation cost of NMN/NR metabolism
- Resveratrol 250–1,000 mg/day — sirtuin-activating compound; fat-soluble, take with a meal containing healthy fat for absorption
- Quercetin 500–1,000 mg/day — senolytic that may reduce the CD38-driven inflammaging that depletes NAD+ in the first place; some protocols use intermittent rather than daily dosing
- Fisetin 100–500 mg/day — alternative or additional senolytic
- Apigenin 50–100 mg/day — CD38 inhibitor that may reduce NAD+ consumption
This is a maximalist stack; many practitioners use only NMN/NR + TMG. The senolytics in particular have substantial individual cost and limited individual evidence, and their inclusion is a matter of risk tolerance and budget. Caloric restriction or time-restricted eating, regular endurance exercise, and adequate sleep do far more for sirtuin activation than any supplement, and should be the foundation on which supplementation is layered.
For the deeper picture of how to combine these interventions, see Longevity Protocols.
Cautions
- Theoretical cancer concern — cancer cells are metabolically active and could in principle benefit from enhanced NAD+ availability. The countervailing argument is that NAD+ also supports DNA repair and immune surveillance, which should be cancer-protective. Until longer human data are available, individuals with active cancer or strong family history should discuss NAD+ supplementation with their oncologist before starting.
- Methylation cofactor depletion — chronic high-dose NMN or NR can deplete methyl donors. Add TMG 500–1,000 mg/day if taking NMN/NR above 500 mg/day chronically. Monitor homocysteine periodically.
- Long-term safety unknown — no NMN trial has run longer than 24 weeks. The biology of any chronic intervention that influences mitochondrial function, sirtuin activity, and epigenetic state at a population level over decades is not yet characterized.
- Quality variation in the supplement market — analytical surveys have found that some commercial NMN products contain less NMN than labeled. Purchase from manufacturers that publish third-party certificates of analysis for purity and potency.
- Pregnancy and breastfeeding — no safety data; avoid.
- Drug interactions — NMN may enhance insulin sensitivity and could potentiate hypoglycemia in patients on insulin or sulfonylureas (see Energy & Mitochondria).
Key Research Papers
- Imai, S., Guarente, L. (2014). NAD+ and Sirtuins in Aging and Disease. Trends in Cell Biology 24(8), 464–471. — DOI
- Bonkowski, M.S., Sinclair, D.A. (2016). Slowing Ageing by Design: The Rise of NAD+ and Sirtuin-Activating Compounds. Nature Reviews Molecular Cell Biology 17(11), 679–690. — DOI
- Mills, K.F., et al. (2016). Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metabolism 24(6), 795–806. — DOI
- Camacho-Pereira, J., et al. (2016). CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction Through a SIRT3-Dependent Mechanism. Cell Metabolism 23(6), 1127–1139. — DOI
- Kanfi, Y., et al. (2012). The Sirtuin SIRT6 Regulates Lifespan in Male Mice. Nature 483(7388), 218–221. — PubMed
- Yoshino, J., Baur, J.A., Imai, S. (2018). NAD+ Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metabolism 27(3), 513–528. — DOI
- Covarrubias, A.J., et al. (2021). NAD+ Metabolism and Its Roles in Cellular Processes During Ageing. Nature Reviews Molecular Cell Biology 22(2), 119–141. — DOI
- Verdin, E. (2015). NAD+ in Aging, Metabolism, and Neurodegeneration. Science 350(6265), 1208–1213. — DOI
- Yoshino, M., et al. (2021). Nicotinamide Mononucleotide Increases Muscle Insulin Sensitivity in Prediabetic Women. Science 372(6547), 1224–1229. — DOI
- Martens, C.R., et al. (2018). Chronic Nicotinamide Riboside Supplementation Is Well-Tolerated and Elevates NAD+ in Healthy Middle-Aged and Older Adults. Nature Communications 9(1), 1286. — DOI
- Gomes, A.P., et al. (2013). Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication During Aging. Cell 155(7), 1624–1638. — PubMed
- Walker, M.A., Bhargava, R. (2025). Clinical Evidence for the Use of NAD+ Precursors to Slow Aging. Geromedicine. — PubMed
PubMed Topic Searches
- PubMed: sirtuin NAD+ aging
- PubMed: Sinclair lab NAD+ and epigenetic reprogramming
- PubMed: NMN lifespan/healthspan
- PubMed: caloric restriction mimetic SIRT1
- PubMed: FOXO-SIRT1 autophagy
Connections
- NAD+ & NMN Overview
- NAD+ Benefits Hub
- NAD+ for Cognition
- NAD+ for Energy & Mitochondria
- NAD Precursors Compared
- Longevity Protocols
- Fasting
- Rapamycin
- Vitamin B3 (Niacin)
- Quercetin (Senolytic)
- Fisetin (Senolytic)
- All Antioxidants
- Methylene Blue
- Alpha Lipoic Acid
- Oxidative Stress
- Cancer (theoretical concern)