Aspartic Acid and Energy Production
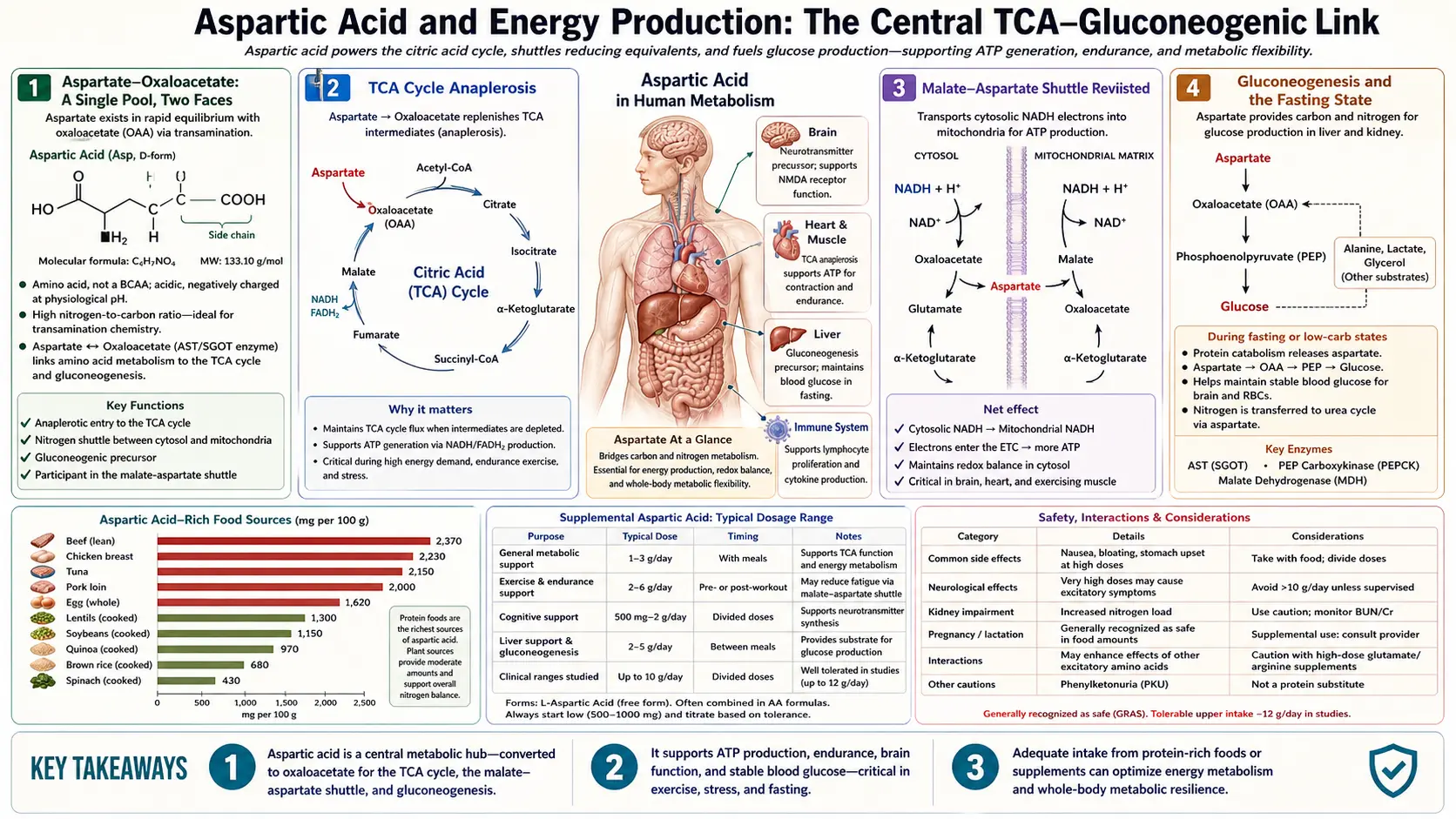
Aspartate sits at a strategic intersection of cellular energy metabolism. Through transamination with oxaloacetate, it is in direct equilibrium with the TCA cycle — aspartate and oxaloacetate are essentially the same metabolic pool seen from two sides. Through the malate-aspartate shuttle, it carries reducing equivalents (NADH) from cytosol to mitochondria, where they fuel oxidative phosphorylation and ATP synthesis. Through gluconeogenesis, it supplies the carbon skeleton for new glucose during fasting. And through the purinosome, it donates nitrogen for adenine and adenosine synthesis — making it indirectly essential for every molecule of ATP the cell produces. This page walks through each role and explains why aspartate has been marketed (with limited evidence) as a fatigue-fighting supplement for over half a century.
Table of Contents
- Aspartate-Oxaloacetate: A Single Pool, Two Faces
- TCA Cycle Anaplerosis
- The Malate-Aspartate Shuttle Revisited
- Gluconeogenesis and the Fasting State
- Purine Synthesis and ATP Building Blocks
- Ammonia, Aspartate, and Exercise-Induced Fatigue
- Potassium-Magnesium Aspartate: The 1960s Fatigue Story
- Modern Evidence on Aspartate Supplementation for Performance
- Aspartate in Mitochondrial Disease and Chronic Fatigue
- Clinical Takeaways
- Key Research Papers
- Connections
- Featured Videos
Aspartate-Oxaloacetate: A Single Pool, Two Faces
The enzyme aspartate aminotransferase (AST, also called glutamic-oxaloacetic transaminase, GOT) catalyzes the freely-reversible transamination:
L-aspartate + alpha-ketoglutarate ↔ oxaloacetate + L-glutamate
The reaction is at near-equilibrium under most cellular conditions, meaning aspartate and oxaloacetate are interconverted on a sub-second timescale and their concentrations are tightly coupled. Practically, this means that the cell does not maintain a meaningful distinction between "aspartate pool" and "oxaloacetate pool" — they are a single metabolic pool of the four-carbon dicarboxylic acid, with nitrogen attached or not depending on local supply and demand.
This coupling has profound consequences. Anywhere oxaloacetate is needed (the citrate synthase step of the TCA cycle, gluconeogenesis, the malate-aspartate shuttle), aspartate is an immediately available substrate via AST. Anywhere aspartate is needed (the urea cycle ASS1 step, purine biosynthesis, asparagine synthesis), oxaloacetate is an immediately available source via AST in reverse. The two pools breathe in unison.
The AST enzyme exists in two isoforms: mitochondrial (mAST) and cytosolic (cAST), with similar kinetic properties but distinct subcellular distribution. Both are released into circulation upon hepatocyte damage and are the AST measured on the routine liver panel — elevated AST signals hepatocellular injury and is one of the most common abnormal liver tests. The clinical interpretation is purely as a damage marker; circulating AST has no measurable functional role.
The dependence of the entire AST reaction on vitamin B6 (as pyridoxal-5-phosphate) is the molecular reason why B6 deficiency can produce subtle metabolic dysfunction across many systems. Without adequate B6, AST capacity drops, the aspartate-oxaloacetate coupling weakens, and downstream pathways (urea cycle, malate-aspartate shuttle, purine synthesis) become rate-limited.
TCA Cycle Anaplerosis
The TCA cycle (also called the Krebs cycle, citric acid cycle, or tricarboxylic acid cycle) is the central catabolic pathway by which acetyl-CoA derived from fats, carbohydrates, and amino acids is oxidized to CO2, generating the reduced cofactors (NADH, FADH2) that drive ATP synthesis at the electron transport chain.
The cycle is composed of eight enzymatic steps that convert oxaloacetate plus acetyl-CoA to two molecules of CO2, regenerating oxaloacetate for the next turn. In principle the cycle is catalytic — oxaloacetate is regenerated each cycle — but in practice TCA cycle intermediates are constantly withdrawn for biosynthetic purposes (citrate exported for fatty acid synthesis, alpha-ketoglutarate diverted to glutamate, oxaloacetate to aspartate and gluconeogenesis, succinyl-CoA to heme synthesis). These cataplerotic withdrawals must be replaced by anaplerotic reactions that resupply cycle intermediates.
Aspartate is one of the major anaplerotic inputs. Via AST, aspartate-derived oxaloacetate refills the TCA cycle:
- During high amino acid catabolism (fasting, high-protein meal, stress), aspartate flow into oxaloacetate maintains TCA cycle flux
- During high gluconeogenic demand, aspartate flow supports the export of oxaloacetate as malate, which drives gluconeogenesis
- During exercise in muscle, aspartate from the purine nucleotide cycle (see below) supplies anaplerotic carbons
The other major anaplerotic input is pyruvate carboxylase, which combines pyruvate plus CO2 plus ATP to make oxaloacetate. This is the dominant anaplerotic pathway in the well-fed state when pyruvate is abundant. The two pathways — aspartate-derived and pyruvate-derived — are complementary, with the relative contribution depending on dietary state and tissue.
The Malate-Aspartate Shuttle Revisited
The malate-aspartate shuttle was described in detail on the Urea Cycle page; here we focus on its energy-production role specifically.
Glycolysis converts glucose to pyruvate in the cytosol, generating 2 molecules of cytosolic NADH per glucose. The NADH must be re-oxidized to NAD+ for glycolysis to continue, but the inner mitochondrial membrane is impermeable to NADH itself. Two shuttle systems address this:
- Malate-aspartate shuttle — the dominant system in heart, liver, kidney, and brain. Delivers NADH equivalents to mitochondrial Complex I (NADH dehydrogenase), generating ~2.5 ATP per cytosolic NADH
- Glycerol-3-phosphate shuttle — the dominant system in skeletal muscle and brown adipose tissue. Delivers NADH equivalents to mitochondrial Complex II / ubiquinone via FADH2, generating ~1.5 ATP per cytosolic NADH
The choice of shuttle has measurable consequences for cellular ATP yield per glucose. A heart cell using the malate-aspartate shuttle gets approximately 32 ATP per glucose; a fast-twitch muscle cell relying on the glycerol-3-phosphate shuttle gets approximately 30 ATP per glucose. The difference is small but multiplied across the chronic energy budget of an organ makes a difference, and explains in part why the heart (which never stops working) cannot tolerate the bioenergetic deficits that would barely affect skeletal muscle.
The shuttle has clinical relevance in two contexts. In diabetic cardiomyopathy, chronic hyperglycemia and metabolic dysregulation impair malate-aspartate shuttle function in cardiomyocytes, contributing to the energy deficit characteristic of diabetic heart disease. In cancer metabolism, the Warburg phenomenon (preferential glycolysis even in the presence of oxygen) shifts the cell's ATP balance toward glycolytic ATP and away from oxidative ATP, reducing dependence on the malate-aspartate shuttle. Many cancer types have characteristic alterations in shuttle expression.
Gluconeogenesis and the Fasting State
During fasting (overnight, prolonged exercise, ketogenic diet, type 1 diabetes between meals), the liver and kidney synthesize new glucose from non-carbohydrate precursors via gluconeogenesis. The major substrates are lactate (from anaerobic glycolysis in red blood cells and exercising muscle), glycerol (from triglyceride breakdown), and the glucogenic amino acids — of which aspartate is one of the most efficient.
The aspartate-to-glucose pathway:
- Aspartate → oxaloacetate (via AST)
- Oxaloacetate → phosphoenolpyruvate (PEP) via PEP carboxykinase (PEPCK), consuming GTP. This is one of the major regulatory steps of gluconeogenesis.
- PEP runs the gluconeogenic pathway in reverse to glycolysis, producing glyceraldehyde-3-phosphate → fructose-1,6-bisphosphate → fructose-6-phosphate → glucose-6-phosphate
- Glucose-6-phosphatase (a liver-and-kidney-specific enzyme) hydrolyzes glucose-6-phosphate to free glucose, which is released into circulation
Each molecule of aspartate consumed yields one half-molecule of glucose (two aspartates produce one glucose). The pathway is most active during overnight fasting and prolonged exercise; chronic high-protein diets that exceed the body's protein needs convert excess amino acid carbon to glucose via this route, contributing to the glucose disposal of dietary protein.
From the energy-production perspective, aspartate's gluconeogenic role means that during fasting, aspartate-derived glucose helps maintain blood glucose homeostasis for brain function (the brain cannot use fatty acids for fuel and depends on glucose plus ketones during fasting). This is a critical role even though aspartate is not itself a direct ATP-producing substrate.
Purine Synthesis and ATP Building Blocks
The purine nucleotides — adenine and guanine derivatives — are the molecular building blocks of DNA, RNA, and the high-energy nucleotide cofactors that drive virtually every cellular reaction: ATP, GTP, NADH, NADPH, FAD, coenzyme A, S-adenosylmethionine, and many others. Without purine nucleotides, no biosynthesis, no signaling, no energy currency.
De novo purine synthesis is a 10-step pathway that builds the purine ring atom-by-atom on a ribose-5-phosphate backbone. Each step incorporates specific nitrogen and carbon atoms from different donors: glycine donates the N7-C8-C5 segment, glutamine donates N9 and N3, formate (via N10-formyl-tetrahydrofolate) donates C2 and C8, CO2 donates C6, and aspartate donates N1.
The aspartate-donation step (step 8 of de novo purine biosynthesis) uses adenylosuccinate synthetase to link aspartate to IMP, forming adenylosuccinate. The next enzyme, adenylosuccinate lyase, cleaves off fumarate, leaving AMP. The released fumarate is recycled back through the TCA cycle, completing a small metabolic loop called the purine nucleotide cycle that is particularly active in exercising skeletal muscle.
The implication for energy production is direct: aspartate is in the construction lineage of every adenine nucleotide. Each ATP molecule the cell synthesizes traces its N1 atom to a molecule of aspartate. The body's enormous ATP turnover (an adult cycles approximately his/her own body weight in ATP per day) makes purine biosynthesis one of the most demanding ongoing metabolic processes, and aspartate is a quantitatively significant input.
The purine nucleotide cycle in exercising muscle deserves separate mention. During intense exercise, muscle AMP rises (from ATP → ADP → AMP), and AMP deaminase converts AMP to IMP plus ammonia. The IMP is reaminated back to AMP using aspartate as the N1 donor, releasing fumarate (anaplerotic input to the TCA cycle) and consuming the ammonia. The net effect is to generate anaplerotic carbons for the TCA cycle from amino acid sources, supporting sustained energy production during exercise. This is one mechanism proposed to underlie the ergogenic claims for aspartate supplementation, though clinical evidence is limited.
Ammonia, Aspartate, and Exercise-Induced Fatigue
Exercise produces ammonia from two sources: the purine nucleotide cycle (described above) and branched-chain amino acid catabolism in working muscle. Ammonia accumulation is one of several mechanisms hypothesized to contribute to muscular and central fatigue during prolonged exercise. The mechanism is multifactorial: ammonia disrupts pH regulation, may interfere with brain neurotransmitter balance (the so-called "central fatigue hypothesis"), and reduces glycolytic flux at high concentrations.
Aspartate's role in ammonia handling provides a mechanistic rationale for its proposed ergogenic effect:
- Aspartate provides nitrogen for the urea cycle, which clears ammonia produced by amino acid catabolism in the liver
- Aspartate provides nitrogen for the purine nucleotide cycle, which can re-incorporate ammonia produced in exercising muscle back into AMP
- Aspartate-derived anaplerotic fumarate supports continued TCA cycle flux during exercise, maintaining oxidative ATP production
The composite hypothesis: aspartate supplementation should delay the onset of exercise-induced fatigue by enhancing ammonia clearance and supporting mitochondrial energy production. This was the foundation of the substantial mid-twentieth-century supplement literature on potassium-magnesium aspartate as a fatigue-fighter.
Potassium-Magnesium Aspartate: The 1960s Fatigue Story
From the late 1950s through the early 1970s, potassium-magnesium aspartate was actively marketed and clinically studied as a fatigue-relieving supplement. The combination products (Spartase, Spasmag, and others, primarily European) typically delivered 1-2 g aspartate per day plus elemental potassium and magnesium. Indications included general fatigue, recovery from surgery or illness, and athletic performance.
Several small clinical trials in the 1960s reported subjective benefit. Rosen et al. (1962) reported reduced perceived exertion at submaximal exercise loads. Wesson et al. (1964) reported delayed time-to-exhaustion in endurance cycling. Several European studies in postoperative recovery reported faster mobilization. The mechanism was attributed to aspartate's role in ammonia clearance and energy production, plus the obvious effect of magnesium and potassium repletion (the patients in these studies were often electrolyte-depleted from their underlying conditions).
Methodologically the early trials were limited — small sample sizes, inconsistent blinding, subjective outcomes — and the supplement-quality and dosing variation was substantial. The category never made it into mainstream sports medicine, and by the 1980s interest had largely waned.
The modern reincarnation has been D-aspartic acid for testosterone (covered on the D-Aspartate page), which is mechanistically distinct from the L-aspartate fatigue claims. The L-aspartate fatigue claim itself has not had a sustained research program, though mineral-aspartate salts (magnesium aspartate, calcium aspartate, zinc aspartate) remain popular forms of mineral supplementation because the aspartate moiety chelates the mineral and enhances bioavailability (compared to oxide or carbonate forms, though typically equivalent to citrate or glycinate forms).
Modern Evidence on Aspartate Supplementation for Performance
The modern sports nutrition consensus is that L-aspartate supplementation provides no measurable ergogenic benefit for trained athletes with adequate nutrition. The mechanistic rationale is real, but the body's endogenous aspartate synthesis (from oxaloacetate, glutamate, and asparagine) is more than sufficient under typical conditions, and exogenous aspartate is metabolized rapidly without a sustained increase in tissue concentration at the relevant sites.
Specific recent findings:
- Meta-analyses of aspartate-containing supplements for exercise performance show no consistent effect on time-to-exhaustion, peak power, or perceived exertion
- Mineral-aspartate salts (Mg-Asp, K-Asp) for electrolyte repletion are well-tolerated and effective but no superior to other bioavailable mineral forms
- Aspartame intake (the artificial sweetener that metabolizes to aspartate, phenylalanine, and methanol) at typical intake levels does not produce measurable ergogenic or performance-detrimental effects in athletes
- Whole-food protein intake from animal or plant sources provides ample aspartate without need for isolated supplementation
The exception to the "no benefit" pattern is in repletion of frank deficiency states — severely malnourished patients in recovery, postoperative patients with poor nutrition, advanced cirrhosis patients with sarcopenia and protein-energy malnutrition. In these populations, comprehensive amino acid repletion (which includes aspartate as part of the total amino acid pool) does improve recovery and functional status. But this is not specifically an aspartate effect — it is a total nutritional rehabilitation effect.
Aspartate in Mitochondrial Disease and Chronic Fatigue
Primary mitochondrial diseases (MELAS, MERRF, Leigh syndrome, mitochondrial DNA depletion syndromes, and many others) are characterized by impaired oxidative phosphorylation and the resulting bioenergetic deficit. Affected tissues are typically those with the highest energy demand: brain, heart, skeletal muscle, kidney, retina, inner ear.
Aspartate metabolism is dysregulated in many mitochondrial diseases. Plasma aspartate concentrations can be elevated (due to impaired downstream utilization), and the alanine:aspartate ratio is a research biomarker in some mitochondrial myopathies. The aralar (mitochondrial aspartate-glutamate antiporter) deficiency described on the Urea Cycle page is itself a recognized mitochondrial disease with severe neurodevelopmental presentation.
Therapeutic implications are limited. Direct aspartate supplementation has not been shown to benefit primary mitochondrial disease patients. The general management uses the "mitochondrial cocktail" of CoQ10, L-carnitine, riboflavin (B2), thiamine (B1), creatine, and alpha-lipoic acid, with variable evidence. Aspartate is not a standard component.
For chronic fatigue syndrome (ME/CFS) and the chronic fatigue overlap with long COVID, fibromyalgia, and POTS, the bioenergetic hypothesis (that affected patients have impaired mitochondrial function in skeletal muscle, brain, or both) is one of several working models. Some clinicians prescribe a similar "mitochondrial cocktail" with mixed evidence, occasionally including aspartate-containing supplements. The clinical effect sizes are modest at best, and the heterogeneity of the patient population makes it difficult to identify which subgroups, if any, benefit. Aspartate alone is not an evidence-based intervention.
Clinical Takeaways
- Aspartate is mechanistically essential to cellular energy production through the TCA cycle, the malate-aspartate shuttle, gluconeogenesis, and purine biosynthesis. It is not optional for life.
- Endogenous synthesis is normally adequate. Aspartate is a non-essential amino acid because the body can make it from oxaloacetate and glutamate. Dietary aspartate from protein-containing foods provides additional supply but is not separately required.
- Isolated aspartate supplementation does not enhance energy or athletic performance in well-nourished healthy adults. The mechanistic rationale is real but the marginal contribution from supplementation is below the threshold of detectable benefit.
- Mineral-aspartate salts (magnesium aspartate, potassium aspartate, zinc aspartate) are reasonable forms of mineral supplementation, comparable in bioavailability to glycinate or citrate forms and superior to oxide or carbonate forms.
- For chronic fatigue, mitochondrial disease, or chronic illness, aspartate is part of comprehensive nutritional support but is not a primary intervention. The evidence does not support isolated aspartate as a fatigue treatment.
- L-ornithine L-aspartate (LOLA) is the one specific clinical context in which exogenous aspartate is administered to drive a metabolic pathway — the urea cycle in hepatic encephalopathy — with modest evidence of efficacy. See the Urea Cycle page.
- Vitamin B6 status matters for aspartate metabolism — AST requires pyridoxal-5-phosphate, and B6 deficiency can impair aspartate handling across multiple pathways. Severe B6 deficiency is uncommon in developed-country populations but worth checking in chronic illness, alcoholism, malabsorption, or pregnancy.
Key Research Papers
- Krebs HA, Johnson WA (1937). The role of citric acid in intermediate metabolism in animal tissues. Enzymologia. The original TCA cycle description. — PubMed
- Owen OE et al. (2002). The key role of anaplerosis and cataplerosis for citric acid cycle function. Journal of Biological Chemistry. — PubMed
- Lu M et al. (2010). The malate-aspartate shuttle. Frontiers in Bioscience. — PubMed
- Birsoy K et al. (2015). An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. — PubMed
- Sullivan LB et al. (2015). Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. — PubMed
- Rosen H, Blumenthal A, McCallum J (1962). Effect of potassium and magnesium aspartates on exercise tolerance. Federation Proceedings. — PubMed
- Wesson M et al. (1988). Effects of oral administration of aspartates on the metabolic response to prolonged exhausting exercise. European Journal of Applied Physiology. — PubMed
- Tullson PC, Terjung RL (1991). Adenine nucleotide metabolism in contracting skeletal muscle. Exercise and Sport Sciences Reviews. The purine nucleotide cycle in exercise. — PubMed
- Banister EW, Cameron BJ (1990). Exercise-induced hyperammonemia: peripheral and central effects. International Journal of Sports Medicine. — PubMed
- Cynober L (2002). Plasma amino acid levels with a note on membrane transport: characteristics, regulation, and metabolic significance. Nutrition. — PubMed
- Owen OE, Kalhan SC, Hanson RW (2002). The key role of anaplerosis and cataplerosis for citric acid cycle function. JBC. — PubMed
- Brand MD (2005). The efficiency and plasticity of mitochondrial energy transduction. Biochemical Society Transactions. — PubMed
PubMed Topic Searches
- PubMed: Malate-aspartate shuttle
- PubMed: Aspartate anaplerosis TCA cycle
- PubMed: Aspartate gluconeogenesis
- PubMed: Purine nucleotide cycle exercise
- PubMed: Mitochondrial disease amino acid therapy
Connections
- Aspartic Acid Benefits Hub
- Aspartic Acid Overview
- Neurotransmission & NMDA
- Aspartate in the Urea Cycle
- D-Aspartate & Testosterone
- Glutamic Acid
- Alanine
- Glycine
- Glutamine
- Magnesium (Mg-Aspartate)
- Potassium (K-Aspartate)
- Vitamin B6 (AST Cofactor)
- Chronic Fatigue
- Mitochondrial Support
- All Amino Acids