Aspartic Acid and the Urea Cycle
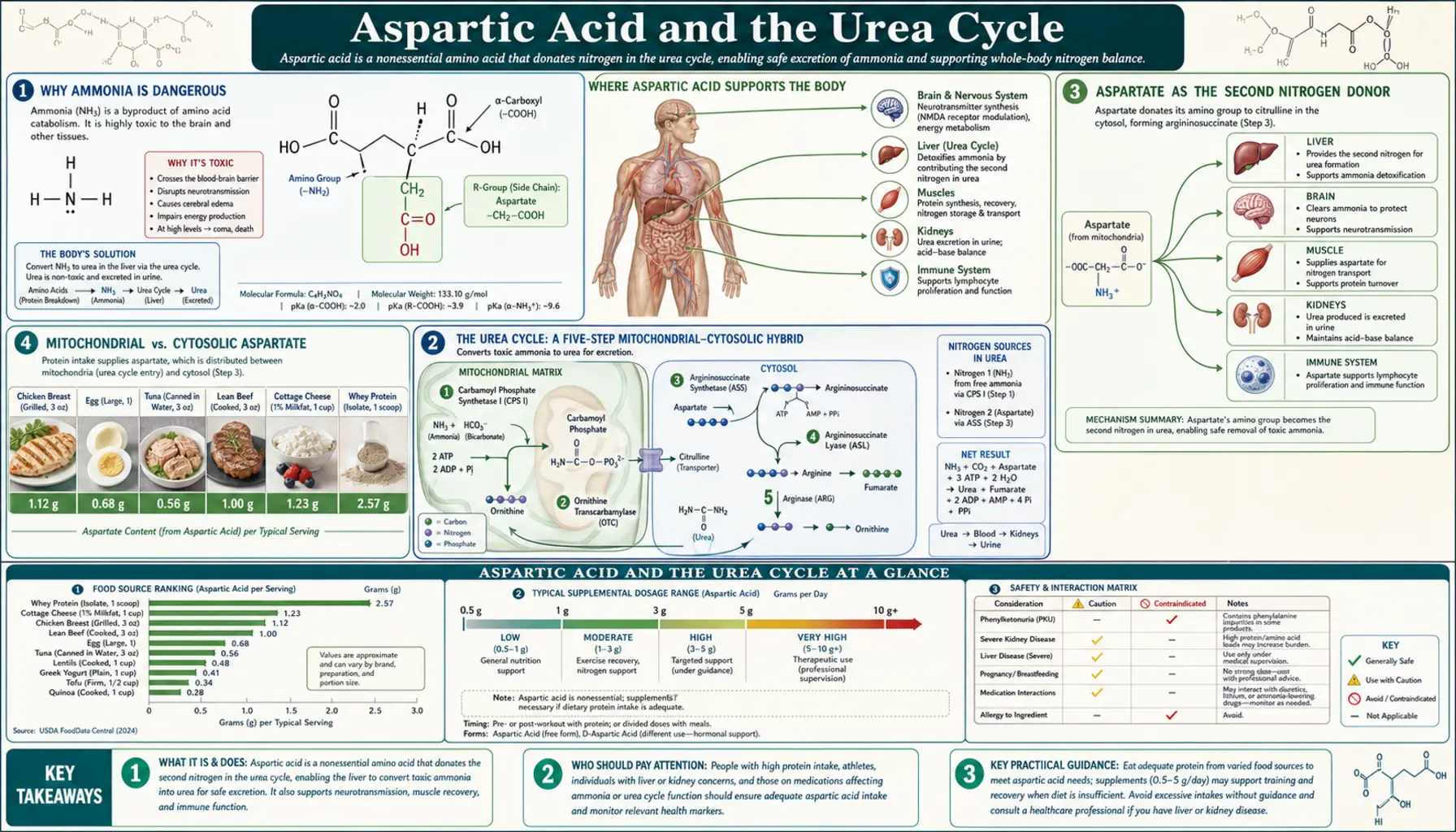
Every gram of protein a person eats generates ammonia as a byproduct of amino acid catabolism — and ammonia at even modest concentrations is profoundly toxic to the brain, producing the disorientation, asterixis, and ultimately coma of hepatic encephalopathy. The urea cycle is the body's principal mechanism for converting this ammonia into urea, an inert nitrogen carrier excreted in urine. Aspartate is one of two nitrogen donors that feed the cycle — the only nitrogen source for the argininosuccinate synthetase step — making it indispensable for safe protein metabolism. The same molecule that powers the urea cycle also bridges the cytosolic and mitochondrial NADH pools through the malate-aspartate shuttle, a kind of biochemical second job that becomes critical in any high-energy tissue. This page walks through both roles, the eight major urea cycle defects, and the clinical management of hyperammonemia.
Table of Contents
- Why Ammonia Is Dangerous
- The Urea Cycle: A Five-Step Mitochondrial-Cytosolic Hybrid
- Aspartate as the Second Nitrogen Donor
- Mitochondrial vs Cytosolic Aspartate
- Urea Cycle Defects: CPS1, OTC, ASS1, ASL, ARG1
- Acute and Chronic Hyperammonemia
- The Malate-Aspartate Shuttle
- Clinical Monitoring: Plasma Ammonia and Amino Acids
- Nutritional Management of Urea Cycle Disorders
- Liver Disease and Acquired Hyperammonemia
- Key Research Papers
- Connections
- Featured Videos
Why Ammonia Is Dangerous
Ammonia (NH3) and its protonated form ammonium (NH4+) are in equilibrium at physiologic pH, with ammonium predominating. At normal blood concentrations (10-35 micromolar in adults, slightly higher in newborns), this poses no problem. Above approximately 100 micromolar, neurologic symptoms appear; above 200 micromolar, cerebral edema and altered consciousness develop; and above 500 micromolar, herniation and death become imminent without urgent intervention.
The neurotoxicity has several mechanisms operating in parallel. Ammonia readily crosses the blood-brain barrier in its uncharged NH3 form. Inside astrocytes, glutamine synthetase incorporates ammonia into glutamate to form glutamine — the body's primary ammonia-handling reaction. This is initially protective, but in sustained hyperammonemia, intracellular glutamine accumulates to osmotically active concentrations, drawing water into astrocytes and producing the cerebral edema that dominates the clinical picture. Glutamine accumulation also disrupts mitochondrial function within the astrocyte itself. Glutamatergic and GABAergic neurotransmission is disrupted by altered substrate availability. The composite effect is the syndrome of hepatic encephalopathy or, in the acute pediatric setting, urea cycle disorder crisis: confusion, asterixis (the flapping hand tremor), hyperventilation (respiratory alkalosis driven by the ammonia itself acting on brainstem chemoreceptors), seizure, and coma.
The therapeutic urgency is high. Neurologic outcome correlates with duration of severe hyperammonemia — even a few hours above 200 micromolar can produce persistent deficits, and pediatric urea cycle disorders presenting in the newborn period have devastating prognoses if not rapidly treated.
The Urea Cycle: A Five-Step Mitochondrial-Cytosolic Hybrid
The urea cycle was the first metabolic cycle ever described — Hans Krebs (the same Krebs of the citric acid cycle) and Kurt Henseleit worked it out in 1932, five years before the citric acid cycle. It operates predominantly in the liver, with smaller activity in kidney and small intestine. The five steps are:
- Carbamoyl phosphate synthetase I (CPS1) — mitochondrial. Combines ammonia (from glutamate via glutamate dehydrogenase) with bicarbonate and 2 ATP to form carbamoyl phosphate. N-acetylglutamate (NAG) is an obligatory allosteric activator. This is the rate-limiting and committed step of the cycle.
- Ornithine transcarbamylase (OTC) — mitochondrial. Combines carbamoyl phosphate with ornithine to produce citrulline. OTC is the most common urea cycle defect, X-linked.
- Argininosuccinate synthetase (ASS1) — cytosolic. Combines citrulline (transported out of the mitochondrion) with aspartate, using 1 ATP, to produce argininosuccinate. This is where aspartate enters the cycle and donates its alpha-amino nitrogen.
- Argininosuccinate lyase (ASL) — cytosolic. Cleaves argininosuccinate into arginine and fumarate. The fumarate is the "exit door" by which the carbon skeleton of aspartate leaves the urea cycle and rejoins the TCA cycle.
- Arginase 1 (ARG1) — cytosolic. Hydrolyzes arginine into urea and ornithine. The urea is exported into the bloodstream for renal excretion; the ornithine is transported back into the mitochondrion to begin the cycle again.
The net stoichiometry: two molecules of ammonia (one from glutamate via CPS1, one from aspartate via ASS1) plus one molecule of CO2 plus three ATP yield one molecule of urea, plus the fumarate that connects to the TCA cycle. Each turn of the cycle removes two nitrogens; the carbon skeleton of aspartate is recovered as fumarate and can be reused.
Aspartate as the Second Nitrogen Donor
The argininosuccinate synthetase (ASS1) step is the only point in the urea cycle where aspartate is required. Aspartate donates its alpha-amino group to citrulline, forming argininosuccinate. The reaction consumes one ATP, hydrolyzed to AMP plus pyrophosphate, the most energetically expensive of the cycle's steps.
The aspartate available for this reaction is generated by transamination of oxaloacetate (an intermediate of the TCA cycle and the gluconeogenic pathway) by aspartate aminotransferase (AST). AST transfers an amino group from glutamate to oxaloacetate, producing alpha-ketoglutarate (which re-enters the TCA cycle) and aspartate (which is consumed by ASS1). This linkage means that:
- The carbon skeleton of urea-cycle aspartate ultimately derives from oxaloacetate (a TCA intermediate)
- The nitrogen of urea-cycle aspartate derives from glutamate (the body's principal amino-nitrogen donor)
- The carbon exits the urea cycle as fumarate from the ASL step, ready to feed back into the TCA cycle
- Aspartate, oxaloacetate, fumarate, and the TCA cycle are tightly integrated with urea synthesis
The integration means that hepatic energy state and urea cycle flux are not independent. Anything that impairs hepatic TCA cycle function (severe hypoxia, mitochondrial poisoning, ATP depletion) will also impair urea cycle function and produce hyperammonemia. Conversely, conditions that increase amino acid catabolism (severe sepsis, major trauma, prolonged starvation with adaptive ketosis, gastrointestinal bleeding into the gut) will increase the demand for urea cycle throughput and may exceed the cycle's capacity, producing a clinical hyperammonemia syndrome even with structurally intact urea cycle enzymes.
Mitochondrial vs Cytosolic Aspartate
Because the urea cycle straddles the mitochondrial-cytosolic boundary (steps 1 and 2 in mitochondria, steps 3-5 in cytosol), the aspartate consumed at the ASS1 step must reach the cytosol. The principal mechanism is the citrin/aralar antiporter system on the inner mitochondrial membrane, which exchanges cytosolic glutamate plus proton for mitochondrial aspartate.
Citrin (encoded by SLC25A13) is the liver-predominant isoform; aralar (SLC25A12) is the brain-predominant isoform. Both perform the same biochemical function: they enable aspartate generated in the mitochondrion (via mitochondrial AST acting on the abundant oxaloacetate of the TCA cycle) to reach the cytosol where ASS1 needs it.
Defects in citrin produce citrullinemia type 2 (CTLN2) in adults — a urea cycle disorder presenting with episodic hyperammonemia, sometimes triggered by alcohol or a high-carbohydrate meal. Patients often have a characteristic aversion to sweet foods and preference for protein, reflecting an unconscious adaptation to their metabolic dysfunction. Treatment includes a low-carbohydrate, protein-balanced diet and arginine supplementation; severe cases may require liver transplantation.
Aralar deficiency produces a much rarer infantile neurologic disorder with global developmental delay, epilepsy, and hypomyelination. The brain version of the antiporter is essential for normal neurodevelopment because brain glutamate-glutamine cycling depends on mitochondrial-cytosolic aspartate flux.
Urea Cycle Defects: CPS1, OTC, ASS1, ASL, ARG1
Each of the five urea cycle enzymes can be deficient as a single-gene disorder. The combined incidence is approximately 1 in 35,000 live births, making urea cycle disorders the most common cause of inherited hyperammonemia. The five primary enzyme defects are:
- CPS1 deficiency — autosomal recessive. Severe neonatal presentation typical. No citrulline or arginine production. Treatment with sodium benzoate, sodium phenylbutyrate, arginine supplementation; severe cases require liver transplant.
- OTC deficiency — X-linked, the most common urea cycle disorder (1 in 14,000 to 1 in 80,000 depending on population). Hemizygous males typically present in the neonatal period with severe disease; heterozygous females have a milder, variable phenotype due to X-inactivation skewing. Elevated urinary orotic acid is the diagnostic biochemical clue (carbamoyl phosphate that cannot enter the urea cycle is shunted into pyrimidine synthesis, producing orotic aciduria). Treatment as for CPS1, plus avoidance of catabolic triggers.
- ASS1 deficiency (citrullinemia type 1) — autosomal recessive. Citrulline cannot accept aspartate. Citrulline accumulates dramatically in plasma (50- to 100-fold elevation) and is the diagnostic biochemical signature. Treatment includes nitrogen scavengers, low-protein diet, and citrulline-free formula in newborns.
- ASL deficiency (argininosuccinic aciduria) — autosomal recessive. Argininosuccinate accumulates and is excreted in urine in large quantities. Patients have a characteristic trichorrhexis nodosa (fragile, brittle hair) and chronic hepatic dysfunction in addition to hyperammonemia episodes.
- Arginase 1 (ARG1) deficiency — autosomal recessive. Unique among urea cycle disorders for typically presenting in childhood (not infancy) with a slowly progressive spastic diplegia rather than acute hyperammonemia crisis. Arginine accumulates in plasma.
Three additional disorders affect cofactors or transporters: NAG synthase (NAGS) deficiency (no allosteric activator for CPS1), HHH syndrome (hyperornithinemia-hyperammonemia-homocitrullinuria, defective ornithine transport into mitochondria), and the citrin deficiency / CTLN2 described above. The combined recognition of these eight as "urea cycle disorders" is standard in inborn-error-of-metabolism textbooks.
From the aspartate perspective, ASS1 deficiency is the most directly relevant — it is the step where aspartate would normally donate its nitrogen, and the resulting citrulline accumulation is the direct biochemical signature. ASL deficiency is the next step in the cycle, where aspartate's carbon skeleton would normally exit as fumarate.
Acute and Chronic Hyperammonemia
Hyperammonemia is a medical emergency at concentrations above 200 micromolar in any patient and above 100 micromolar in a neonate. The clinical presentation depends on how rapidly the ammonia rises:
- Acute hyperammonemia (urea cycle crisis) — rapid onset over hours. Vomiting, lethargy, hyperventilation (respiratory alkalosis), seizures, coma. In neonates the only sign may be progressive somnolence and poor feeding — clinically indistinguishable from sepsis in early stages, which is why neonatal screening with plasma ammonia is critical in any unwell neonate.
- Chronic hyperammonemia — weeks to months. Failure to thrive, developmental delay, episodic vomiting, protein aversion. Common in partial enzyme deficiencies (heterozygous female OTC, for example).
- Acquired hyperammonemia — occurs in cirrhosis (hepatic encephalopathy), portosystemic shunts, valproic acid use, urinary diversion procedures, gastrointestinal hemorrhage, and severe protein overload. Mechanism is reduced hepatic urea cycle capacity rather than primary enzyme defect.
Acute management focuses on three pillars:
- Stop the nitrogen input. NPO; dextrose-containing IV fluids to suppress endogenous protein catabolism; lipid to provide non-nitrogenous calories.
- Provide alternative nitrogen excretion routes. Sodium benzoate combines with glycine to form hippurate (renally excreted). Sodium phenylacetate or phenylbutyrate combines with glutamine to form phenylacetylglutamine (renally excreted). The combination product Ammonul (sodium benzoate + sodium phenylacetate) is the standard IV agent.
- Hemodialysis or hemofiltration if ammonia exceeds 500 micromolar or rises despite scavenger therapy. Continuous renal replacement therapy is often the first choice in neonates.
Arginine supplementation is also typically given because all urea cycle defects except arginase deficiency render arginine an "essential" amino acid (since the urea cycle is the body's principal route of arginine synthesis). Without supplemental arginine, urea cycle disorder patients become severely arginine-deficient.
The Malate-Aspartate Shuttle
Beyond the urea cycle, aspartate's second great metabolic role is in the malate-aspartate shuttle — the principal mechanism by which cytosolic NADH (generated during glycolysis) is transported across the inner mitochondrial membrane to be oxidized by the electron transport chain. The inner mitochondrial membrane is impermeable to NADH itself, so a shuttle is required.
The cycle works as follows:
- Cytosolic NADH reduces oxaloacetate to malate (cytosolic malate dehydrogenase)
- Malate enters the mitochondrion via the dicarboxylate antiporter (exchanging for alpha-ketoglutarate)
- Inside the mitochondrion, malate is oxidized back to oxaloacetate (mitochondrial malate dehydrogenase), regenerating NADH inside the mitochondrial matrix where it can feed Complex I of the electron transport chain
- Mitochondrial oxaloacetate accepts an amino group from glutamate (mitochondrial AST) to form aspartate plus alpha-ketoglutarate
- Aspartate exits the mitochondrion via the citrin/aralar antiporter (exchanging for cytosolic glutamate plus a proton)
- Cytosolic aspartate is transaminated back to oxaloacetate (cytosolic AST), completing the cycle
The net effect: cytosolic NADH equivalents are transferred to the mitochondrial matrix without the membrane having to be permeable to NADH itself. The shuttle is highly efficient (each cytosolic NADH yields the same ~2.5 ATP as a directly mitochondrial NADH) and dominates in tissues with high oxidative metabolism: liver, heart, kidney, and brain.
Tissues that rely instead on the alternative glycerol-3-phosphate shuttle (skeletal muscle, brown adipose tissue) lose energy efficiency because that shuttle delivers electrons to FAD rather than NAD, generating only 1.5 ATP per cytosolic NADH. The choice of shuttle is one reason why heart muscle (malate-aspartate dominant) is more energy-efficient per glucose molecule than fast-twitch skeletal muscle (glycerol-3-phosphate dominant).
Clinical Monitoring: Plasma Ammonia and Amino Acids
For any patient with suspected hyperammonemia or known urea cycle disorder, the laboratory workup includes:
- Plasma ammonia — the central measurement. Must be processed quickly (within 30 minutes of draw) on ice, as ammonia rises in vitro due to red cell amino acid metabolism. Normal: 10-35 micromolar (adults), up to 110 micromolar (newborns)
- Quantitative plasma amino acids — identifies the specific urea cycle defect. Elevated citrulline = ASS1 or ASL deficiency. Low citrulline = CPS1 or OTC. Elevated arginine = arginase. Elevated argininosuccinate = ASL.
- Urinary orotic acid — elevated in OTC deficiency and HHH syndrome (because carbamoyl phosphate shunts into pyrimidine synthesis), normal in CPS1 deficiency. The most useful single test for distinguishing CPS1 from OTC.
- Liver function tests, blood gas, glucose, lactate — to exclude acquired causes (hepatic failure, organic acidemia, severe sepsis)
- Plasma aspartate — not typically used in clinical workup, but research interest in elevated plasma aspartate as a marker of hepatic ASS1 deficiency or as a flux marker
For chronic management of known urea cycle disorder patients, monitoring includes plasma ammonia, plasma amino acids (to titrate dietary protein), serum albumin, plasma essential amino acids (often deficient on protein-restricted diets), growth parameters, and neurodevelopmental assessment.
Nutritional Management of Urea Cycle Disorders
Long-term management of urea cycle disorders rests on three pillars: protein restriction to the minimum needed for growth, supplementation with the specific deficient downstream amino acid (typically arginine), and continuous low-dose nitrogen scavenger therapy.
Protein restriction. Total protein intake is reduced to the minimum compatible with normal growth in children (typically 1.0-1.5 g/kg/day instead of the usual 1.5-2.0 g/kg/day for healthy infants) and roughly 0.6-0.8 g/kg/day in adults. Protein is provided as a mix of "natural" intact protein (containing essential amino acids the body cannot make) plus an "essential amino acid only" medical formula. This minimizes nitrogen load while preserving essential amino acid intake.
Arginine or citrulline supplementation. Patients with proximal defects (CPS1, OTC) cannot make arginine; supplementation prevents arginine deficiency. Patients with distal defects (ASS1, ASL) can be given citrulline instead, which is more efficiently retained because each molecule consumed donates an extra ammonia molecule per cycle turn.
Continuous nitrogen scavenger therapy. Oral sodium phenylbutyrate (Buphenyl) or glycerol phenylbutyrate (Ravicti) is taken three to four times daily. These convert to phenylacetate in vivo, conjugate with glutamine in the liver and kidney, and excrete in urine as phenylacetylglutamine, providing an "alternate pathway" for nitrogen disposal.
From the aspartate perspective, no specific aspartate supplementation is recommended for urea cycle disorder patients. The body normally synthesizes adequate aspartate from oxaloacetate via AST, and additional dietary aspartate would simply add to the nitrogen load the cycle is trying to dispose of.
Liver Disease and Acquired Hyperammonemia
The most common adult cause of hyperammonemia is not a urea cycle defect — it is acquired hepatic dysfunction. Cirrhosis, acute liver failure, and surgically or congenitally created portosystemic shunts all reduce the liver's effective urea-cycle capacity by either destroying hepatocyte mass or bypassing the portal venous delivery of ammonia to the liver.
The clinical syndrome is hepatic encephalopathy (HE), ranging from subclinical cognitive deficits (Stage 0/1 on the West Haven criteria) to deep coma (Stage 4). Asterixis is the classical examination finding. Plasma ammonia is elevated but the correlation with clinical grade is imperfect; the diagnosis is clinical.
Treatment of HE focuses on reducing gut-derived ammonia rather than enhancing urea cycle capacity:
- Lactulose — an indigestible disaccharide that is fermented by colonic bacteria to short-chain fatty acids, lowering colonic pH. The lower pH traps ammonia as ammonium in the colon, preventing absorption. Also produces an osmotic diarrhea that reduces gut transit time and bacterial nitrogen production.
- Rifaximin — a poorly absorbed antibiotic that reduces colonic bacterial nitrogen production. Combined with lactulose for refractory HE.
- Branched-chain amino acid (BCAA) supplementation — oral BCAAs (leucine, isoleucine, valine) can improve cognition in some HE patients and may modestly extend survival in cirrhosis. The mechanism involves restoration of the BCAA:aromatic amino acid ratio, which is disrupted by cirrhosis.
- L-ornithine L-aspartate (LOLA) — an intravenous formulation directly delivering aspartate plus ornithine to enhance urea cycle flux and ammonia clearance. Approved for HE in many European countries; used off-label in the US. Modest evidence of efficacy in acute HE.
The LOLA formulation is the one clinical context in which exogenous aspartate is administered specifically to support urea cycle function. The pharmacologic logic is that providing extra aspartate (along with ornithine, the substrate for OTC and the starting point of the cycle) drives flux through the impaired hepatic urea cycle. The clinical effect size is modest but the safety profile is excellent.
Key Research Papers
- Krebs HA, Henseleit K (1932). Untersuchungen über die Harnstoffbildung im Tierkörper. Hoppe-Seyler's Zeitschrift. The original urea cycle description. — PubMed
- Häberle J et al. (2019). Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet Journal of Rare Diseases. — PubMed
- Batshaw ML et al. (2014). A longitudinal study of urea cycle disorders. Molecular Genetics and Metabolism. — PubMed
- Saheki T, Kobayashi K (2002). Mitochondrial aspartate-glutamate carrier (citrin) deficiency. Journal of Human Genetics. — PubMed
- Wakabayashi Y et al. (1991). Argininosuccinate synthetase gene structure and chromosomal mapping. Genomics. — PubMed
- Kido J et al. (2019). Long-term outcome and intervention of urea cycle disorders. Journal of Pediatrics. — PubMed
- Brusilow SW et al. (1980). Treatment of episodic hyperammonemia in children with inborn errors of urea synthesis. NEJM. The original sodium benzoate/phenylacetate work. — PubMed
- Stadler S et al. (2014). Newborn screening for urea cycle disorders. European Journal of Pediatrics. — PubMed
- Kircher SG et al. (2017). L-ornithine L-aspartate (LOLA) for hepatic encephalopathy meta-analysis. Hepatology. — PubMed
- Bachmann C (2002). Mechanisms of hyperammonemia. Clinical Chemistry and Laboratory Medicine. — PubMed
- Vilstrup H et al. (2014). Hepatic encephalopathy in chronic liver disease: AASLD/EASL practice guideline. Hepatology. — PubMed
- LaBuda CJ, Brusilow SW (2009). Carbamoyl phosphate synthetase 1 deficiency review. Pediatric Neurology. — PubMed
PubMed Topic Searches
- PubMed: Urea cycle disorder aspartate
- PubMed: Hyperammonemia management
- PubMed: Argininosuccinate synthetase citrullinemia
- PubMed: Malate-aspartate shuttle
- PubMed: Citrin deficiency CTLN2
Connections
- Aspartic Acid Benefits Hub
- Aspartic Acid Overview
- Neurotransmission & NMDA
- D-Aspartate & Testosterone
- Aspartate and Energy Production
- Arginine
- Ornithine
- Citrulline
- Glutamic Acid
- Glutamine
- Liver Disease
- Cirrhosis
- Hepatic Encephalopathy
- Inborn Errors of Metabolism
- All Amino Acids