aPTT Test: Activated Partial Thromboplastin Time
The activated Partial Thromboplastin Time (aPTT) measures the speed and integrity of the intrinsic coagulation pathway — the route triggered by contact activation with negatively charged surfaces inside blood vessels. It is the primary test for monitoring unfractionated heparin (UFH) therapy, diagnosing hemophilia A and B, and detecting the lupus anticoagulant. Understanding aPTT requires grasping three concepts: the intrinsic pathway cascade, the difference between factor deficiency and inhibitors, and why the same prolonged aPTT can mean bleeding risk in one patient and thrombotic risk in another.
Table of Contents
- Overview — Intrinsic Coagulation Pathway
- How the aPTT Is Measured
- Normal Range and Therapeutic Targets
- Heparin Monitoring with aPTT
- Unfractionated vs. Low-Molecular-Weight Heparin
- Prolonged aPTT — Differential Diagnosis
- The Mixing Study — Factor Deficiency vs. Inhibitor
- Hemophilia A and B
- Lupus Anticoagulant and Antiphospholipid Syndrome
- Key Research and Citations
- Connections
- Featured Videos
Overview — Intrinsic Coagulation Pathway
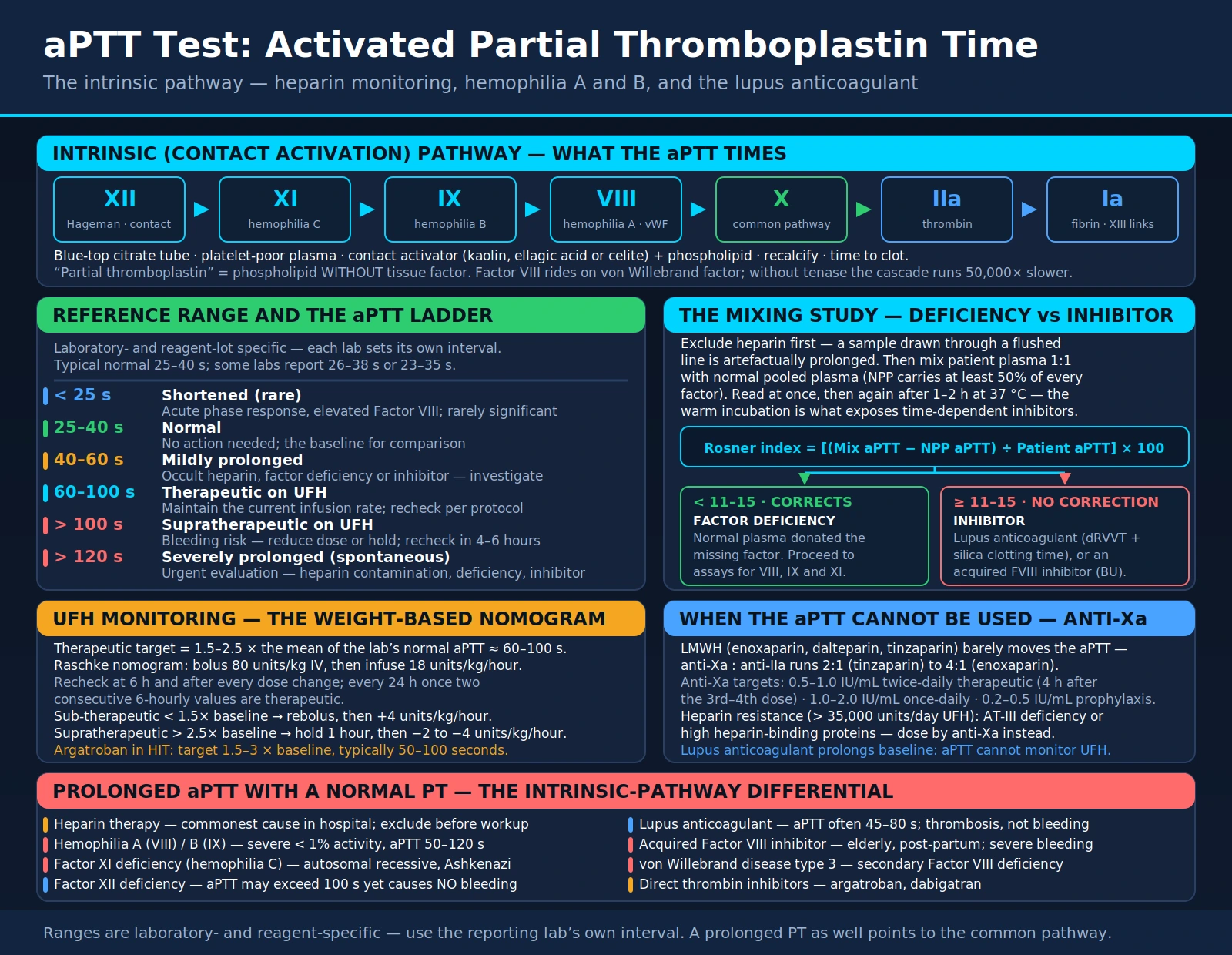
The intrinsic (contact activation) pathway begins when Factor XII (Hageman factor) binds to negatively charged surfaces such as collagen, polyphosphates, kallikrein, or laboratory glass. This triggers a sequential activation cascade: XII → XI → IX → VIII (cofactor) → X. The activated Factor X, combined with Factor Va (the prothrombinase complex), converts prothrombin to thrombin, which cleaves fibrinogen to fibrin. This final fibrin mesh, cross-linked by Factor XIII, constitutes the stable clot that arrests hemorrhage.
The key cofactor in this pathway is Factor VIII, which circulates in the bloodstream bound to von Willebrand factor (vWF). VWF serves as a carrier protein that protects Factor VIII from premature degradation. When Factor IXa is generated, it forms the tenase complex on a phospholipid surface with Factor VIIIa and calcium, dramatically accelerating Factor X activation. Without this amplification step, the intrinsic pathway proceeds 50,000 times more slowly — a clinically disastrous inefficiency that explains why even partial Factor VIII or IX deficiency can cause life-threatening bleeding.
Deficiency of Factor VIII causes Hemophilia A; deficiency of Factor IX causes Hemophilia B (Christmas disease). Both are X-linked recessive disorders — the genes for both factors reside on the X chromosome — so these conditions predominantly affect males, who carry only one X chromosome and therefore have no functional backup copy. Females with one defective copy are typically carriers with mildly reduced factor levels that rarely cause spontaneous bleeding. Both hemophilias prolong the aPTT while leaving the prothrombin time (PT) completely normal, because the extrinsic pathway (measured by PT) uses tissue factor to activate Factor VII, bypassing the entire intrinsic cascade.
The intrinsic pathway converges with the extrinsic pathway at Factor X activation — the so-called "common pathway." Factors X, V, II (prothrombin), and fibrinogen are shared by both pathways. Severe deficiencies of any of these common pathway factors will therefore prolong both the PT and aPTT simultaneously, a pattern that helps clinicians narrow the differential diagnosis rapidly. Understanding this anatomy of the coagulation cascade is essential for correctly interpreting every abnormal aPTT result.
How the aPTT Is Measured
Blood is drawn into a citrate (blue-top) tube. Sodium citrate chelates ionized calcium, the essential cofactor for virtually every coagulation enzyme. This calcium chelation immediately halts coagulation in the collection tube, preserving the sample for accurate measurement. The blue-top tube must be filled to the correct volume — under-filling dilutes the citrate ratio and artificially prolongs clotting times, while over-filling reduces the citrate concentration and may allow partial activation.
In the laboratory, platelet-poor plasma is separated by centrifugation and then incubated with a contact activator. The contact activator — kaolin (a silicate clay), ellagic acid, or celite (diatomaceous earth) — provides the negatively charged surface that triggers Factor XII activation, initiating the intrinsic cascade. Phospholipid (a surrogate for platelet membranes) is added to support the tenase and prothrombinase complexes. This is the "partial thromboplastin" component of the test name: phospholipid alone, without tissue factor. Complete thromboplastin — used in the PT — contains both phospholipid and tissue factor, activating the extrinsic pathway.
After incubation, the plasma mixture is recalcified with calcium chloride. A photometric detector or mechanical wire detects clot formation; the elapsed time from calcium addition to clot formation is reported as the aPTT in seconds. The "activated" part of the name refers specifically to the addition of the contact activator, which standardizes Factor XII activation and makes results reproducible across laboratories.
The aPTT is substantially more sensitive to heparin than the PT because heparin enhances antithrombin's inhibition of thrombin and Factors IXa, Xa, XIa, and XIIa — all enzymes in the intrinsic and common pathways. At therapeutic intravenous heparin levels, the aPTT will typically double or more relative to the patient's baseline. The PT, by contrast, reflects the extrinsic pathway and is relatively insensitive to heparin except at very high concentrations.
Preanalytical variables that affect aPTT accuracy include: insufficient blood volume in the collection tube, prolonged transport time (>4 hours at room temperature), improper centrifugation leaving residual platelets, hemolysis or lipemia, and extremely elevated hematocrit (requiring citrate volume adjustment in polycythemia). Clinicians should repeat any unexpected result with a freshly drawn sample before making major therapeutic decisions.
Normal Range and Therapeutic Targets
The normal aPTT range varies by laboratory and reagent lot — each institution must establish its own reference interval based on its specific contact activator and instrumentation. A typical normal range is 25–40 seconds, but some laboratories report 26–38 seconds or 23–35 seconds. Always interpret aPTT results against the reporting laboratory's established reference range, not a textbook number.
The therapeutic target for unfractionated heparin (UFH) monitoring is defined relative to the laboratory's own reference range rather than an absolute second value. Current guidelines recommend targeting 1.5–2.5 times the mean of the institution's normal aPTT range, which commonly corresponds to approximately 60–100 seconds.
| aPTT Range | Interpretation | Clinical Action |
|---|---|---|
| < 25 seconds | Shortened (rare) | May indicate acute phase response, elevated Factor VIII; rarely clinically significant alone |
| 25–40 seconds | Normal | No action needed; baseline for comparison |
| 40–60 seconds | Mildly prolonged | Investigate cause; check for occult heparin, factor deficiency, or inhibitor |
| 60–100 seconds | Therapeutic (on UFH) | Maintain current heparin infusion rate; recheck per protocol |
| > 100 seconds on UFH | Supratherapeutic | Increased bleeding risk; reduce dose or hold infusion; recheck in 4–6 hours |
| > 120 seconds (spontaneous) | Severely prolonged | Urgent evaluation; rule out heparin contamination, severe deficiency, or inhibitor |
For hemophilia management, the aPTT is not used to set a therapeutic target — factor levels are measured directly by chromogenic or one-stage clotting assays. For argatroban monitoring in heparin-induced thrombocytopenia (HIT), the target aPTT is 1.5–3 times baseline (typically 50–100 seconds). For bivalirudin, aPTT targeting also applies in certain protocols, though activated clotting time (ACT) is used in procedural settings.
Heparin Monitoring with aPTT
Unfractionated heparin (UFH) is a heterogeneous mixture of glycosaminoglycan chains ranging from 3,000 to 30,000 daltons in molecular weight. Its primary mechanism is the activation of antithrombin III (AT-III), a serine protease inhibitor that constitutively inhibits thrombin and several other coagulation factors at a slow baseline rate. When heparin binds to a specific pentasaccharide sequence on antithrombin, it induces a conformational change that accelerates AT-III's inhibitory activity by approximately 1,000-fold. The heparin-AT complex then inactivates thrombin (Factor IIa) and Factor Xa most potently, and also inhibits Factors IXa, XIa, and XIIa.
Because UFH has highly variable pharmacokinetics — it binds to plasma proteins (histidine-rich glycoprotein, vitronectin, fibronectin, platelet factor 4), endothelial cells, and macrophages to different degrees in different patients — its anticoagulant effect cannot be predicted from the dose alone. Laboratory monitoring is therefore essential. The aPTT has been the standard monitoring test for UFH for decades.
The standard weight-based heparin nomogram (Raschke et al., 1993) is the foundation of modern UFH dosing:
- Check baseline aPTT (and CBC, PT) before initiating heparin.
- Initiate with a weight-based bolus (typically 80 units/kg IV) followed by a continuous infusion (typically 18 units/kg/hour).
- Check aPTT 6 hours after initiation and after every dose adjustment.
- Once therapeutic aPTT is achieved on two consecutive measurements 6 hours apart, transition to checking every 24 hours.
- If aPTT is sub-therapeutic (< 1.5× baseline), administer a repeat bolus and increase the infusion rate by 4 units/kg/hour.
- If aPTT is supratherapeutic (> 2.5× baseline), hold the infusion for 1 hour and reduce the rate by 2–4 units/kg/hour.
Heparin resistance is defined as a failure to achieve therapeutic aPTT despite high UFH doses (typically > 35,000 units/day). Causes include antithrombin III deficiency (congenital or acquired — as in nephrotic syndrome, DIC, or post-cardiac bypass), elevated heparin-binding proteins (acute phase proteins rise in inflammation), and technical issues with assay reagents. In heparin resistance, anti-Xa levels provide a more accurate measure of heparin effect and should be used to guide dosing.
Heparin-induced thrombocytopenia (HIT) is a severe immunological complication in which antibodies against the heparin-PF4 complex activate platelets, causing paradoxical thrombosis. HIT reduces platelet count by >50% from baseline, typically 5–10 days after heparin initiation. When HIT is suspected, heparin must be stopped immediately and a non-heparin anticoagulant (argatroban, bivalirudin, fondaparinux) substituted. The aPTT remains useful for monitoring argatroban in HIT.
Unfractionated vs. Low-Molecular-Weight Heparin
Low-molecular-weight heparins (LMWHs) — including enoxaparin, dalteparin, and tinzaparin — are produced by chemical or enzymatic depolymerization of UFH, yielding chains averaging 4,000–6,500 daltons. The shorter chain length fundamentally changes their pharmacology: the minimum pentasaccharide sequence needed to bind antithrombin is preserved, but the additional length needed to simultaneously bind thrombin (Factor IIa) is lost in most molecules. As a result, LMWHs predominantly inhibit Factor Xa (anti-Xa) over thrombin, with anti-Xa:anti-IIa ratios ranging from approximately 2:1 (tinzaparin) to 4:1 (enoxaparin).
This pharmacological shift has important practical consequences. Because LMWHs bind fewer plasma proteins and have more predictable subcutaneous bioavailability (~90% vs. ~30% for UFH), they have predictable dose-response relationships that make routine aPTT monitoring unnecessary in most patients. Standard dosing for venous thromboembolism treatment is 1 mg/kg enoxaparin subcutaneously every 12 hours (or 1.5 mg/kg once daily) without any laboratory monitoring in patients with normal renal function and body weight.
The aPTT is unreliable for LMWH monitoring. At therapeutic LMWH doses, the aPTT may be only minimally elevated or entirely normal, because the degree of thrombin inhibition (which drives aPTT prolongation) is insufficient to lengthen clotting time detectably. Attempting to use aPTT to confirm LMWH therapeutic effect will generate falsely reassuring results and is not recommended.
Anti-Xa monitoring IS indicated for LMWH in specific high-risk populations:
- Renal impairment (creatinine clearance < 30 mL/min): LMWH is renally cleared; accumulation increases bleeding risk
- Obesity (BMI > 40 kg/m²): weight-based dosing may be inaccurate at extremes
- Pregnancy: volume of distribution changes with gestational age; anti-Xa monitoring guides dose adjustment
- Extremes of weight: both very low (< 50 kg) and very high (> 120 kg) body weights
- Pediatrics: variable pharmacokinetics in neonates and infants
Target anti-Xa levels for LMWH: 0.5–1.0 IU/mL for twice-daily therapeutic dosing (measured 4 hours after the third or fourth dose); 1.0–2.0 IU/mL for once-daily dosing. Prophylactic dosing targets 0.2–0.5 IU/mL. Fondaparinux, a synthetic pentasaccharide that selectively and irreversibly inhibits Factor Xa alone, also does not prolong the aPTT at any therapeutic concentration and is monitored by anti-Xa if needed.
Prolonged aPTT — Differential Diagnosis
A prolonged aPTT is defined as an aPTT above the laboratory's reference range (typically > 40 seconds). The clinical context — whether the patient is on anticoagulation, has a personal or family bleeding history, or has a thrombotic event — is essential for correct interpretation. The pattern of aPTT abnormality in relation to PT narrows the differential considerably.
Isolated Prolonged aPTT (PT Normal)
This pattern indicates a defect confined to the intrinsic pathway (Factors XII, XI, IX, VIII) or the presence of a direct inhibitor of intrinsic pathway factors or phospholipids:
- Heparin therapy: the most common cause in hospitalized patients; always exclude before extensive workup
- Hemophilia A (Factor VIII deficiency): X-linked; lifelong bleeding history; aPTT often 60–100+ seconds in severe disease
- Hemophilia B (Factor IX deficiency, Christmas disease): clinically identical to Hemophilia A; distinguished by factor assay
- Factor XI deficiency (Hemophilia C): autosomal recessive; most common in Ashkenazi Jewish populations; variable and often mild bleeding phenotype not closely correlated with factor level
- Factor XII deficiency: paradoxically, does NOT cause bleeding — Factor XII is not essential for in vivo hemostasis; patients with severe Factor XII deficiency have a normal bleeding history and may actually have elevated thrombosis risk; aPTT may be dramatically prolonged (> 100 seconds)
- Lupus anticoagulant / antiphospholipid syndrome: an inhibitor that interferes with phospholipid-dependent in vitro assays; causes in vivo thrombosis, not bleeding
- Von Willebrand disease, severe type 3: complete VWF deficiency causes secondary Factor VIII deficiency (VIII circulates bound to VWF); type 3 is the only vWD subtype that significantly prolongs aPTT
- Direct thrombin inhibitors (argatroban, dabigatran): prolong aPTT and thrombin time (TT); dabigatran elevates aPTT at therapeutic concentrations
- Acquired Factor VIII inhibitor: autoantibody; usually in elderly, post-partum women, or patients with lymphoma/solid tumors; can cause severe spontaneous bleeding
Both PT and aPTT Prolonged
This pattern suggests a common pathway defect, multi-factor deficiency, or a global anticoagulant effect:
- Severe liver disease or hepatic failure: liver synthesizes Factors I, II, V, VII, IX, X, XI; severe dysfunction impairs synthesis of all vitamin K-dependent and non-vitamin K-dependent factors
- Disseminated intravascular coagulation (DIC): consumptive coagulopathy depletes multiple factors simultaneously; thrombocytopenia and elevated D-dimer accompany the coagulopathy
- Warfarin or vitamin K deficiency: primarily prolongs PT (affects Factors II, VII, IX, X), but at supratherapeutic levels also prolongs aPTT
- Massive transfusion or dilutional coagulopathy: large-volume blood product replacement without adequate factor replacement
- Common pathway factor deficiency: rare isolated deficiency of Factor X, V, II, or fibrinogen (afibrinogenemia/hypofibrinogenemia)
- Amyloidosis: Factor X adsorption by amyloid fibrils; also Factor IX in some forms
The Mixing Study — Factor Deficiency vs. Inhibitor
When an unexpectedly prolonged aPTT is discovered, the first priority is excluding heparin contamination — particularly from heparin flushes in indwelling catheters. If the specimen was drawn through a heparin-containing line, the "prolonged aPTT" may be entirely artifactual. Resampling from a peripheral vein or the contralateral limb resolves this rapidly. If heparin is excluded, the clinically critical next step is a mixing study.
The mixing study principle is elegant: normal pooled plasma contains at least 50% of normal levels of every coagulation factor. If patient plasma is diluted 1:1 with normal plasma:
- If the prolonged aPTT is due to a factor deficiency, the normal plasma donates the missing factor(s), restoring function — the aPTT normalizes or corrects significantly.
- If the prolonged aPTT is due to an inhibitor (an antibody or anticoagulant), the inhibitor remains active in the mix and continues to prolong the aPTT — correction fails.
Protocol:
- Mix patient plasma with normal pooled plasma (NPP) in a 1:1 ratio.
- Measure aPTT immediately on the mix (immediate mix).
- Incubate the mix at 37°C for 1–2 hours, then remeasure (warm incubation mix).
- Compare both results to the aPTT of NPP alone and the patient's aPTT alone.
The warm incubation step is critical for detecting time-dependent inhibitors — particularly the acquired Factor VIII inhibitor, which is an IgG antibody that reacts slowly at 37°C and may not cause non-correction on the immediate mix but clearly fails to correct after incubation. Lupus anticoagulant, by contrast, typically shows non-correction on both immediate and incubated mixes.
Quantifying correction: the Rosner index (also called the Index of Circulating Anticoagulant, ICA) provides an objective cutpoint:
Rosner index = [(Mix aPTT − NPP aPTT) ÷ Patient aPTT] × 100
- Rosner index < 11–15: correction → factor deficiency; proceed to individual factor assays (VIII, IX, XI)
- Rosner index ≥ 11–15: non-correction → inhibitor; proceed to inhibitor identification
Two major inhibitor categories:
- Lupus anticoagulant (LA): most common inhibitor causing prolonged aPTT; non-specific phospholipid-dependent antibody; paradoxically causes thrombosis in vivo; diagnosis requires a panel of tests including dilute Russell's viper venom time (dRVVT), silica clotting time (SCT), and phospholipid neutralization step (Staclot-LA or hexagonal-phase phospholipid test)
- Acquired Factor VIII inhibitor: specific IgG autoantibody against Factor VIII; presents with acute severe bleeding in a patient with no prior bleeding history; Factor VIII level profoundly low; Bethesda assay quantifies inhibitor titer (Bethesda units, BU); managed with bypassing agents — recombinant activated Factor VII (rFVIIa, NovoSeven) or activated prothrombin complex concentrate (aPCC, FEIBA)
Hemophilia A and B
Hemophilia A (Factor VIII deficiency) and Hemophilia B (Factor IX deficiency) are the two most common severe inherited bleeding disorders in males, with a combined prevalence of approximately 1 in 5,000–10,000 live male births. Both are caused by mutations in genes on the X chromosome (Factor VIII gene: Xq28; Factor IX gene: Xq27.1), and both follow X-linked recessive inheritance. Approximately 30% of new cases arise from de novo mutations with no family history.
Severity Classification
Severity correlates with residual clotting factor activity and determines bleeding phenotype:
| Severity | Factor Activity | Clinical Manifestations |
|---|---|---|
| Severe | < 1% (< 0.01 IU/mL) | Spontaneous hemarthrosis, muscle hematomas, life-threatening intracranial bleeding; 2–3 bleeding episodes/month without prophylaxis |
| Moderate | 1–5% (0.01–0.05 IU/mL) | Bleeding with mild to moderate trauma; occasional spontaneous episodes; joint disease develops over decades |
| Mild | 5–40% (0.05–0.40 IU/mL) | Bleeding with significant trauma or surgery; may not be diagnosed until adulthood; normal life expectancy without complications |
Laboratory Pattern
In both Hemophilia A and B:
- aPTT: prolonged (50–120 seconds in severe disease; often normal or marginally prolonged in mild disease)
- PT/INR: normal (extrinsic pathway unaffected)
- Platelet count: normal
- Thrombin time: normal
- Fibrinogen: normal
- Factor VIII level: reduced in Hemophilia A; normal in Hemophilia B
- Factor IX level: reduced in Hemophilia B; normal in Hemophilia A
- Mixing study: corrects (factor deficiency pattern), unless inhibitor has developed
Treatment Advances
Hemophilia A treatment has transformed dramatically:
- Recombinant Factor VIII concentrates: standard prophylaxis (3×/week IV infusions for severe disease)
- Emicizumab (Hemlibra): a bispecific monoclonal antibody that bridges Factor IXa and Factor X, mimicking Factor VIIIa activity; administered subcutaneously (weekly, biweekly, or monthly); dramatically reduces treatment burden; approved for Hemophilia A with or without inhibitors
- Gene therapy: SPK-8011 and other AAV-based Factor VIII gene therapies have achieved sustained factor expression in clinical trials; fitedabio (fidanacogene elaparvovec) was FDA-approved in 2024 for Hemophilia B gene therapy
Inhibitor development — the formation of alloantibodies against infused factor concentrate — affects 25–30% of patients with severe Hemophilia A and approximately 3–5% of those with Hemophilia B. Inhibitors are identified by the Bethesda assay and quantified in Bethesda units (BU). High-titer inhibitors (> 5 BU) require immune tolerance induction (ITI) therapy (prolonged high-dose factor infusions to tolerize the immune system) and bypassing agents for acute bleeds.
Lupus Anticoagulant and Antiphospholipid Syndrome
The lupus anticoagulant (LA) is one of the most clinically treacherous results in coagulation testing: it prolongs clotting tests in vitro but causes thrombosis in vivo — the exact opposite of what the name and lab result might suggest. The name derives from the observation that it was first characterized in patients with systemic lupus erythematosus (SLE), but only 50% of patients with a lupus anticoagulant actually have SLE. The other 50% have antiphospholipid antibody syndrome occurring as a primary disorder, drug-induced (hydralazine, procainamide, chlorpromazine), or associated with infection, malignancy, or other autoimmune conditions.
The lupus anticoagulant is a member of the broader antiphospholipid antibody (aPL) family, which also includes:
- Anticardiolipin antibodies (aCL): IgG or IgM antibodies against the phospholipid cardiolipin; measured by ELISA; medium-high titer IgG most clinically significant
- Anti-β2-glycoprotein I (anti-β2GPI) antibodies: IgG or IgM; β2GPI is a phospholipid-binding protein that serves as the actual antigenic target for most aPL; high specificity for true APS
The in vitro mechanism of LA prolongation: LA antibodies bind to phospholipid-binding proteins (primarily β2GPI) sitting on phospholipid surfaces. This blocks the phospholipid platform required for the tenase and prothrombinase complexes in coagulation assays, slowing the cascade — prolonging aPTT and other phospholipid-dependent tests. Adding excess phospholipid "outcompetes" the LA antibody and normalizes clotting time, which is the basis of phospholipid neutralization confirmatory tests.
The in vivo mechanism of thrombosis: LA antibodies activate endothelial cells (upregulating tissue factor and adhesion molecules), activate platelets, and impair protein C pathway anticoagulation. The net effect is a prothrombotic state causing venous thromboembolism (DVT, pulmonary embolism, cerebral venous thrombosis) and arterial thrombosis (stroke, MI, limb ischemia).
Diagnosis of Antiphospholipid Syndrome
The revised Sapporo/Sydney criteria (Miyakis et al., 2006) require at least one clinical criterion AND at least one laboratory criterion confirmed on two occasions ≥ 12 weeks apart:
- Clinical criteria: (1) vascular thrombosis (any vessel, any organ, confirmed by imaging or histopathology); (2) obstetric morbidity (≥3 unexplained consecutive miscarriages before week 10; ≥1 unexplained fetal death at ≥10 weeks; ≥1 premature birth ≤34 weeks due to severe pre-eclampsia, eclampsia, or placental insufficiency)
- Laboratory criteria: LA detected by phospholipid-dependent coagulation tests; or medium-to-high titer anticardiolipin antibodies; or anti-β2GPI antibodies of IgG or IgM isotype
aPTT in APS Diagnosis and Monitoring
The aPTT is typically prolonged in LA-positive patients, often 45–80 seconds. A key practical consequence: the aPTT CANNOT be used to monitor UFH therapy in patients with LA — the baseline is already prolonged. Anti-Xa levels must be used for UFH monitoring in these patients. Additionally, the aPTT should not be the sole screening test for LA; the International Society on Thrombosis and Haemostasis (ISTH) requires at least two different phospholipid-dependent assays (typically dRVVT and aPTT-based silica clotting time) with confirmatory phospholipid neutralization steps.
Treatment
Long-term anticoagulation is the cornerstone of APS management:
- Venous APS: warfarin with INR target 2.0–3.0 is standard; evidence for higher INR (3.0–4.0) is not convincingly superior for most patients
- Arterial APS (stroke/TIA): combination aspirin + warfarin (INR 2–3) often recommended; debate continues
- Triple-positive APS (all three aPL positive): highest thrombosis recurrence risk; direct oral anticoagulants (DOACs) have shown inferior outcomes vs. warfarin in triple-positive patients and are generally avoided; warfarin preferred
- Obstetric APS without prior thrombosis: low-dose aspirin + prophylactic LMWH during pregnancy and 6 weeks postpartum
- Hydroxychloroquine: adjunctive agent with immunomodulatory and antithrombotic properties; used for APS in SLE patients
Key Research and Citations
- Raschke RA, Reilly BM, Guidry JR, Fontana JR, Srinivas S (1993). The weight-based heparin dosing nomogram compared with a "standard care" nomogram: a randomized controlled trial. Ann Intern Med. 119(9):874–881 — Search PubMed
- Garcia DA, Baglin TP, Weitz JI, Samama MM; American College of Chest Physicians (2012). Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed. Chest. 141(2 Suppl):e24S–e43S. PMID: 22315264
- Miyakis S, Lockshin MD, Atsumi T, et al. (2006). International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 4(2):295–306. PMID: 16420554
- Kershaw G, Orellana D (2013). Mixing tests: diagnostic aides in the investigation of prolonged prothrombin times and activated partial thromboplastin times. Semin Thromb Hemost. 39(3):283–290 — Search PubMed
- Lippi G, Salvagno GL, Montagnana M, Lima-Oliveira G, Guidi GC, Favaloro EJ (2012). Interference of blood cell count parameters on hemostasis testing. Semin Thromb Hemost. 38(6):579–587 — Search PubMed
- Shen YM, Frenkel EP (2004). Thrombosis and a lupus-like anticoagulant in a patient with essential thrombocythemia. Am J Hematol. 75(4):240–242 — Search PubMed
- Meijer P, Kluft C, Kitchen S, Preston FE (2007). The value of laboratory tests for the surveillance of patients on anticoagulant treatment. J Clin Pathol. 60(7):731–734 — Search PubMed
- Konkle BA (2011). Diagnosis and management of von Willebrand disease: basic science to clinical practice. J Thromb Haemost. 9 Suppl 1:346–350 — Search PubMed
- Lusher JM (2000). Inhibitor antibodies to factor VIII and factor IX: management. Semin Thromb Hemost. 26(2):179–188 — Search PubMed
- Ginsberg JS (1996). Management of venous thromboembolism. N Engl J Med. 335(24):1816–1828 — Search PubMed
- Favaloro EJ (2012). Diagnosing von Willebrand disease: a short history of laboratory milestones and a few remaining challenges. Semin Thromb Hemost. 38(7):693–712 — Search PubMed
- Moake JL (2002). Thrombotic microangiopathies. N Engl J Med. 347(8):589–600 — Search PubMed
PubMed Topic Searches
- aPTT and heparin monitoring — PubMed
- Lupus anticoagulant and antiphospholipid syndrome — PubMed
- Hemophilia and Factor VIII inhibitor treatment — PubMed
- Mixing study for coagulation inhibitor vs. factor deficiency — PubMed
Connections
- All Lab Tests
- Coagulation Panel
- PT/INR (Prothrombin Time)
- D-Dimer
- Hemophilia
- Complete Blood Count (CBC)
- Inflammatory Markers
- Cardiovascular Disease