Sarcoidosis: History and Discovery
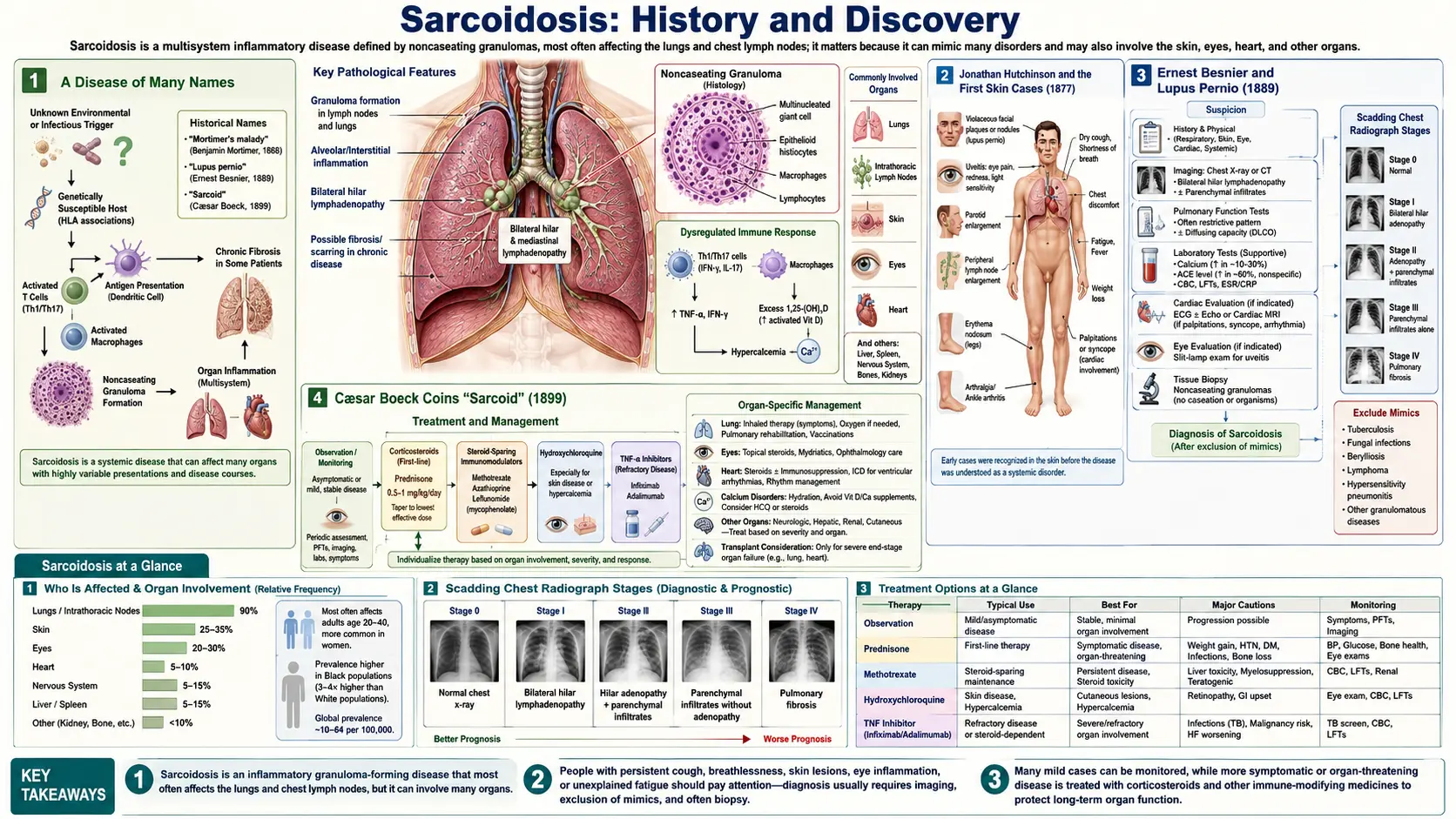
Sarcoidosis is a multisystem disease of unknown cause, defined by the formation of noncaseating granulomas — small, tightly organized clusters of immune cells — in the lungs, lymph nodes, skin, eyes, and many other organs. Its history is unusual because no single person “discovered” it. Instead, several physicians, working in different countries and different decades, each described a piece of the puzzle, which is why the disease was long known by the cumbersome eponym Besnier–Boeck–Schaumann disease. The skin lesions came first, the systemic nature came later, and the cause has still not been found. This page traces that long road of partial discoveries, names the people who walked it, and is careful to separate what is firmly established from what remains, to this day, a genuine medical mystery.
Table of Contents
- A Disease of Many Names
- Jonathan Hutchinson and the First Skin Cases (1877)
- Ernest Besnier and Lupus Pernio (1889)
- Cæsar Boeck Coins “Sarcoid” (1899)
- Jörgen Schaumann Recognizes a Systemic Disease
- The Noncaseating Granuloma and the Tuberculosis Question
- The Kveim–Siltzbach Test and Löfgren Syndrome
- The Cause That Is Still Unknown
- Legacy and the Modern Era
- Research Papers and References
- Connections
- Featured Videos
A Disease of Many Names
Few diseases carry as long a string of historical names as sarcoidosis, and the reason is buried in the way it was discovered. Because its skin signs, its bone changes, its lung disease, and its systemic character were each recognized separately — by different physicians, in different countries, across more than half a century — the condition accumulated a series of eponyms before anyone understood it was all one disease. The most durable of these was Besnier–Boeck–Schaumann disease, honoring the Frenchman Ernest Besnier, the Norwegian Cæsar Boeck, and the Swede Jörgen Schaumann. Other historical labels included Boeck’s sarcoid, Boeck’s disease, Schaumann’s disease, and lymphogranulomatosis benigna.
The name that finally stuck, sarcoidosis, descends from a single word coined by Boeck in 1899 — sarkoid, meaning “flesh-like” or “resembling sarcoma.” That choice of word has caused confusion for over a century, so it is worth stating plainly at the outset: sarcoidosis is not a cancer. Boeck thought the microscopic tissue resembled sarcoma (a malignant tumor of connective tissue), and named it accordingly, but he was describing an appearance, not a diagnosis. The granulomas of sarcoidosis are benign immune structures, not malignant growths. The misleading echo of “sarcoma” inside “sarcoidosis” is a historical accident of nineteenth-century microscopy, nothing more.
The story that follows is therefore not a single discovery but a relay race. Each describer handed the next a clearer view of the same elusive illness, and the modern unified concept of sarcoidosis — one disease, many organs, one characteristic granuloma, one unknown trigger — only came together in the twentieth century. Reading the history in order makes the modern picture far easier to understand.
Jonathan Hutchinson and the First Skin Cases (1877)
The recorded history of sarcoidosis begins with Sir Jonathan Hutchinson (1828–1913), an extraordinarily prolific English surgeon and dermatologist who has often been called “the first sarcoidologist.” In 1877, in his Illustrations of Clinical Surgery, Hutchinson published what is generally regarded as the first illustrated description of the cutaneous disease — raised, dusky-red, painless patches on the skin of the face, arms, and hands. The lesions did not ulcerate and behaved unlike anything in the standard catalogue of skin disease, and Hutchinson recorded the appearance carefully with the colored plates for which his clinical atlases were famous.
Hutchinson’s great contribution was recognizing that these lesions were not tuberculosis and not ordinary lupus — a crucial distinction, because nearly every chronic granular skin disease of the era was being lumped under “lupus” or assumed to be tubercular. He returned to the disease in later writings, and in the 1890s he described a memorable patient, a woman named Mrs. Mortimer, whose dusky purple plaques on the face and forearms he characterized as a distinct entity. In his 1898 account he named her condition “Mortimer’s malady” — one of the earliest attempts to treat these skin findings as a disease in their own right rather than a variant of something else.
It is fair to credit Hutchinson with the opening observation of the entire field, while being honest about its limits: he saw the skin disease clearly, but he did not grasp that it was a systemic condition, nor did he have the microscopic picture that would later define it. His role was that of the careful clinician who first said, in effect, “this is something new.” Everything that followed built on that.
Ernest Besnier and Lupus Pernio (1889)
The next piece came from Paris. In 1889, the French dermatologist Ernest Henri Besnier (1831–1909) described a striking, chronic, violet-colored swelling of the nose, ears, cheeks, and fingers, for which he coined the term lupus pernio (“pernio” refers to chilblain-like, frostbitten-looking skin). The name has endured: lupus pernio remains the term used today for the distinctive disfiguring facial sarcoidosis of the nose and cheeks, and despite its name it is neither lupus erythematosus nor frostbite. It is one of the most recognizable cutaneous signs of the disease.
Besnier was one of the leading dermatologists of his era and his description was precise enough that lupus pernio became a fixed clinical landmark. At the time, however, he too was looking at what appeared to be a localized, peculiar skin condition. The connection between Hutchinson’s patches, Besnier’s lupus pernio, and the disease’s deeper involvement of the lungs and other organs had not yet been drawn. Besnier had added a second well-defined skin manifestation to the record, and his name would eventually take first place in the eponym Besnier–Boeck–Schaumann disease.
What is important historically is that two careful European dermatologists, working independently, had now documented two characteristic skin presentations of the same underlying illness — without either of them knowing it was the same illness, and without knowing what it actually was. The microscope was about to enter the story.
Cæsar Boeck Coins “Sarcoid” (1899)
The word that named the disease came from Norway. In December 1899, Cæsar Peter Møller Boeck (1845–1917), professor of dermatology in Kristiania (now Oslo), published a paper titled Multiple Benign Sarkoid of the Skin. Boeck had done what Hutchinson and Besnier had not: he examined the affected tissue under the microscope. He saw compact collections of epithelioid cells and giant cells — what we now recognize as the granulomas of sarcoidosis — and thought the tissue looked like sarcoma. From the Greek root for “flesh,” he coined the term sarkoid, meaning “flesh-like” or “sarcoma-resembling.”
Here the historical record demands a clear correction that matters for every patient who reads the diagnosis today. Boeck did not believe the disease was cancer, and it is not cancer. He deliberately called it “multiple benign sarkoid” — benign was part of the name — precisely to signal that, although the tissue resembled sarcoma microscopically, it behaved as a harmless growth, not a malignant tumor. The term he created has unfortunately frightened generations of patients who hear “sarc-” and think “sarcoma.” The truth is the opposite of alarming: the resemblance is only skin-deep, the granulomas are immune structures, and sarcoidosis is not a form of cancer.
Boeck’s contribution was therefore twofold. He gave the disease the microscopic identity — the granuloma — that would become its defining feature, and he gave it the root word that, lengthened to sarcoidosis, would eventually become its accepted name. Notably, in later work around 1916 Boeck himself came to appreciate that the condition was not confined to the skin, foreshadowing the systemic understanding that his Swedish contemporary would establish.
Jörgen Schaumann Recognizes a Systemic Disease
The decisive conceptual leap — that all of this was one disease affecting the whole body — is credited chiefly to the Swedish dermatologist Jörgen Nilsen Schaumann (1879–1953). In a body of work carried out in the years around the First World War (his key contributions are usually dated to roughly 1914–1917, with influential publications continuing into the 1930s), Schaumann demonstrated that the skin lesions Besnier had called lupus pernio, the tissue Boeck had called sarcoid, and lesions in the lymph nodes, lungs, bones, tonsils, and other internal organs were all manifestations of a single systemic disease.
Schaumann proposed the name lymphogranulomatosis benigna (benign lymphogranulomatosis) to capture this generalized, granuloma-forming, but non-malignant character — choosing “benign” explicitly to set it apart from the malignant lymphogranulomatosis of Hodgkin disease. His insight reframed everything that had come before: the disparate skin findings of the nineteenth century were not separate curiosities but windows onto a deeper, body-wide process. For this unifying work his name was added to the eponym, completing Besnier–Boeck–Schaumann disease. His name also survives in the Schaumann bodies, the calcium-and-protein inclusions seen inside the giant cells of sarcoid granulomas.
Readers will encounter slightly different years for Schaumann’s key papers across sources — some cite 1914, others 1916 or 1917 — partly because his work appeared over several years and some of it was delayed in publication. What is not in dispute is the substance of his contribution: he is the figure who established that sarcoidosis is a systemic, multi-organ disease, which is the modern definition.
The Noncaseating Granuloma and the Tuberculosis Question
To understand why sarcoidosis was so hard to pin down, it helps to understand the one question that haunted it for decades: is this tuberculosis? Tuberculosis was the great granulomatous disease of the era, and its granulomas look superficially similar to those of sarcoidosis. Distinguishing the two became the central diagnostic problem, and the answer lay in a single microscopic detail.
The pathological hallmark of sarcoidosis is the noncaseating granuloma. A granuloma is a tight, organized ball of immune cells — epithelioid macrophages and multinucleated giant cells ringed by lymphocytes — that the body builds to wall off something it cannot digest. In tuberculosis, the center of the granuloma typically dies and breaks down into a soft, cheese-like (Latin caseum, “cheese”) mass — this is caseating necrosis. In sarcoidosis, classically, the granuloma stays intact: there is no central cheesy necrosis. That single distinction — necrosis present versus absent — became the pathologist’s key to telling the two apart, and the noncaseating granuloma remains the central defining feature of sarcoidosis to this day.
This is also why sarcoidosis is fundamentally a diagnosis of exclusion: because the granulomas of tuberculosis, certain fungal infections, berylliosis, and other conditions can mimic it, a confident diagnosis requires both finding noncaseating granulomas and ruling out the infections and exposures that can produce identical-looking tissue. The historical struggle to separate sarcoidosis from tuberculosis is not a quaint relic; it is built into how the disease is diagnosed even now.
The Kveim–Siltzbach Test and Löfgren Syndrome
The mid-twentieth century added two historically important pieces. The first was a diagnostic test of remarkable peculiarity. In 1941, the Norwegian pathologist Morten Ansgar Kveim (1892–1966) reported that injecting a suspension of tissue taken from the lymph nodes of a sarcoidosis patient into the skin of another sarcoidosis patient would, over several weeks, raise a small noncaseating granuloma at the injection site — in effect, the disease reproducing itself on demand. In the 1950s the American physician Louis Siltzbach refined and standardized the procedure (using spleen-derived material and validating a common reagent internationally), and it became known as the Kveim–Siltzbach test.
For decades this was one of the few reasonably specific tests for sarcoidosis, and it is fascinating historically because it implied that whatever caused the disease was transmissible within the injected tissue — a clue to the still-unknown trigger. The test has since been almost entirely abandoned in practice, because preparing safe, standardized human-tissue reagent is impractical and raises infection and safety concerns; it is now of historical interest rather than routine use, supplanted by tissue biopsy and imaging. But it remains a striking chapter in the disease’s story.
The second piece was the recognition of a distinct acute form. In 1953, the Swedish physician Sven Löfgren (1910–1978) described a characteristic acute presentation — the sudden combination of erythema nodosum (tender red nodules, usually on the shins), bilateral hilar lymphadenopathy (swollen lymph nodes at the roots of both lungs, visible on chest X-ray), often with fever and ankle arthritis. This constellation is now called Löfgren syndrome, and it carries good news: it usually has an excellent prognosis, frequently resolving on its own within one to two years. Recognizing it allowed physicians to distinguish a benign, self-limiting form of sarcoidosis from the chronic, progressive disease — an important practical and prognostic distinction.
The Cause That Is Still Unknown
For all the names, dates, and discoveries above, the central fact of sarcoidosis has never changed and must be stated honestly: its cause remains unknown. More than 140 years after Hutchinson’s first plate, no one has identified what triggers the disease. This is not an evasion or an old-fashioned gap that newer textbooks have quietly filled — current authoritative sources, including recent reviews and national health bodies, continue to classify sarcoidosis as a disease of unknown etiology. Any page claiming a settled cause would be misinforming you.
What is widely accepted is a framework, not an answer. The leading hypothesis — and it should be read as a hypothesis — is that sarcoidosis results from an exaggerated immune response (a vigorous T-helper-cell–driven reaction) to some unidentified trigger or antigen, occurring in people with a genetic predisposition. In this model, a susceptible immune system meets an unknown stimulus and over-responds, building granulomas in an attempt to wall off something it never fully clears. The granuloma, in other words, is the visible footprint of an immune reaction whose original target has never been caught.
Researchers have pursued many candidate triggers without confirming any. Infectious suspects have included mycobacteria (the family that includes the tuberculosis bacterium) and the skin bacterium Cutibacterium (formerly Propionibacterium) acnes; environmental and occupational exposures such as inorganic dusts, silica, mold, and certain insecticides have also been studied, alongside a well-documented genetic component involving HLA immune-system genes. It is essential to be precise here: these are associations and hypotheses under investigation, not proven causes. No single infectious agent or exposure has been established as the cause of sarcoidosis, and most experts suspect the truth may involve several different triggers converging on the same immune pathway. The honest summary — the one this page commits to — is that the cause is genuinely not yet known.
Legacy and the Modern Era
The relay of discovery did not end in Scandinavia. The disease’s modern understanding was shaped by twentieth-century clinicians and epidemiologists who studied it on a large scale, and one feature they documented repeatedly deserves emphasis: sarcoidosis does not strike all populations equally. In the United States it is recognized to occur more frequently, and often more severely, in African Americans than in white Americans, and it shows distinctive patterns across other populations worldwide. Whatever the unknown trigger is, it appears to interact with genetic background — consistent with the susceptibility-plus-trigger hypothesis — and this disparity remains an active and important area of study.
The long eponym Besnier–Boeck–Schaumann disease has gradually given way in everyday medicine to the simpler sarcoidosis, but the three names embedded in the older term are a useful reminder of how the disease was understood piece by piece: Besnier’s lupus pernio on the skin, Boeck’s benign sarcoid under the microscope, and Schaumann’s recognition that it was all one systemic illness — with Hutchinson’s very first observation standing before them all. Each saw a true part; none saw the whole; together they built it.
Today sarcoidosis is approached as a multisystem granulomatous disease diagnosed by combining clinical picture, imaging, and tissue biopsy showing noncaseating granulomas, after excluding mimics. Treatment, when needed, centers on corticosteroids and other immune-modulating drugs aimed at calming the over-active immune response — treating the granuloma-forming reaction even though its original trigger is still unidentified. The history of sarcoidosis is, in the end, a story about the limits and the honesty of medical knowledge: a disease we can name, describe in fine microscopic detail, diagnose, and treat — yet still cannot fully explain. For the clinical picture, organ involvement, and management, see the main Sarcoidosis page.
Research Papers and References
The references below combine historical accounts of the men who described sarcoidosis with current peer-reviewed reviews of its still-unknown cause and immunopathogenesis. Where a stable DOI or PubMed identifier was confirmed it is linked directly; otherwise a curated PubMed topic-search link is provided. Each link opens in a new tab at PubMed or the publisher (National Library of Medicine).
- James DG, Sharma OP. From Hutchinson to now: a historical glimpse. Current Opinion in Pulmonary Medicine. 2002;8(5):416-423. — PubMed: 12172446
- Sharma OP. Sir Jonathan Hutchinson (1828–1913), the first sarcoidologist: a historical sketch. Sarcoidosis, Vasculitis and Diffuse Lung Diseases. 2005. — PubMed: 19382526
- Mortimer’s malady and Hutchinson’s early cutaneous descriptions of sarcoidosis (historical review). PubMed: Hutchinson & Mortimer’s malady
- Ten Berge B, et al. Multiple benign sarkoid of the skin — 100 years since Cæsar Boeck’s pioneering article. — PubMed: 10827505
- Cæsar Boeck and Boeck’s sarcoid (historical biography). — PubMed: 11875931
- James DG. Descriptive definition and historic aspects of sarcoidosis. Clinics in Chest Medicine. 1997;18(4):663-679. — Search PubMed
- Löfgren S. Primary pulmonary sarcoidosis (clinical course and the acute syndrome). Acta Medica Scandinavica. 1953. PubMed: Löfgren 1953
- History and current status of the Kveim–Siltzbach test in sarcoidosis. — PubMed: 7623490
- Statement on Sarcoidosis (ATS/ERS/WASOG joint consensus statement). American Journal of Respiratory and Critical Care Medicine. 1999;160(2):736-755. — doi:10.1164/ajrccm.160.2.ats4-99
- Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. New England Journal of Medicine. 2007;357(21):2153-2165. — doi:10.1056/NEJMra071714
- Grunewald J, et al. Sarcoidosis. Nature Reviews Disease Primers. 2019;5(1):45. — doi:10.1038/s41572-019-0096-x
- Sarcoidosis: etiology, immunopathogenesis, and the search for the unknown trigger (recent reviews). PubMed: sarcoidosis etiology & immunopathogenesis
- Infectious agents (mycobacteria, Cutibacterium/Propionibacterium acnes) and sarcoidosis — systematic reviews. PubMed: sarcoidosis & infectious agents
- Racial and geographic differences in sarcoidosis incidence and severity. PubMed: sarcoidosis epidemiology & race
External Authoritative Resources
- NHLBI (NIH) — Sarcoidosis
- StatPearls (NCBI Bookshelf) — Sarcoidosis
- PubMed — All research on the history of sarcoidosis
Connections
- Pulmonology
- Sarcoidosis (main page)
- All Conditions
- Interstitial Lung Disease
- Tuberculosis
- Pulmonary Hypertension
- Lymphoma
- Lupus